
123
Nicolaus Kröger
John Gribben
Christian Chabannon
Ibrahim Yakoub-Agha
Hermann Einsele
Editors
The EBMT/EHA
CAR-T Cell
Handbook

The EBMT/EHA CAR-T Cell Handbook

Nicolaus Kröger • John Gribben
Christian Chabannon • Ibrahim Yakoub-Agha
Hermann Einsele
Editors
The EBMT/EHA CAR-T Cell
Handbook

Editors
Nicolaus Kröger
Department of Stem Cell Transplantation
University Medical Center
Hamburg-Eppendorf
Hamburg, Germany
Christian Chabannon
Institut Paoli-Calmettes Comprehensive
Cancer Center
Aix-Marseille Université School of
Medicine
Marseille, France
Hermann Einsele
Department of Internal Medicine II
University Hospital Würzburg
Würzburg, Bayern, Germany
John Gribben
Bart’s Cancer Institute
Queen Mary University of London
London, UK
Ibrahim Yakoub-Agha
Maladies du Sang
Unité de Thérapie Cellulaire
Centre hospitalier-Universitaire de Lille
Lille, France
ISBN 978-3-030-94352-3 ISBN 978-3-030-94353-0 (eBook)
https://doi.org/10.1007/978-3-030-94353-0
© The Editor(s) (if applicable) and The Author(s) 2022 This book is an open access publication.
Open Access This book is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit
to the original author(s) and the source, provide a link to the Creative Commons license and indicate if
changes were made.
The images or other third party material in this book are included in the book's Creative Commons
license, unless indicated otherwise in a credit line to the material. If material is not included in the book's
Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the
permitted use, you will need to obtain permission directly from the copyright holder.
The use of general descriptive names, registered names, trademarks, service marks, etc. in this publication
does not imply, even in the absence of a specic statement, that such names are exempt from the relevant
protective laws and regulations and therefore free for general use.
The publisher, the authors and the editors are safe to assume that the advice and information in this book
are believed to be true and accurate at the date of publication. Neither the publisher nor the authors or the
editors give a warranty, expressed or implied, with respect to the material contained herein or for any
errors or omissions that may have been made. The publisher remains neutral with regard to jurisdictional
claims in published maps and institutional afliations.
This Springer imprint is published by the registered company Springer Nature Switzerland AG
The registered company address is: Gewerbestrasse 11, 6330 Cham, Switzerland

v
Preface
Chimeric antigen receptor T cell therapy (CAR-T) is a new class of medicinal prod-
ucts that are genetically engineered from T cells. It is expected that other forms of
Immune Effector Cells-based therapies will soon reach the market, manufactured
from other subsets of immune cells, and engineered through other technologies than
currently used defective retroviral or lentiviral vectors. Cell-based immunotherapies
add to the broader eld of immunotherapies, now populated with monoclonal anti-
bodies including immune checkpoint inhibitors, immune-conjugates, and bi- and
tri-specic antibodies. Approximately 30years after the rst publications reporting
on the development of genetically engineered T cells, expression of a rst genera-
tion chimeric antigen receptor (CAR), and the demonstration of its capability to
recognize antigens in the absence of MHC presentation in the 1980s, the rst com-
mercially available CAR-T cell medicinal products were approved by the FDA and
later by the EMA for the treatment of relapsed/refractory diffuse large B cell lym-
phoma and relapsed/refractory acute lymphoblastic leukemia.
To foster CAR-T cell development and patients’ access to these novel cellular
therapies in Europe, the European Society of Blood and Marrow Transplantation
(EBMT) and the European Hematology Association (EHA) combined forces from
2018—the year of the rst approvals of CAR-T Cells in Europe—by working
closely together in the elds of education, scientic developments, and communica-
tion with health authority and all other stakeholders. It started with the organization
in 2019 of the rst and immediately successful edition of an annual and jointly
organized European CAR-T cell meeting that has become the premier event in the
eld on the European continent. Beyond this major educational initiative, the two
continental professional associations have established the “GoCART-Coalition”
that aims to provide a neutral ecosystem that allows the many interested parties to
communicate and commonly search to solve the many hurdles that the eld is fac-
ing to fully exploit the medical value of these innovative therapies.
In line with this collaboration, the EBMT/EHA Handbook “CAR-T cell ther-
apy”—of which you read the rst edition—was developed. The aim of this hand-
book is to provide the state-of-the-art information on ongoing scientic developments
and medical practices in the eld of CAR-T cell therapies, to enhance knowledge
and practice skills for all categories of healthcare professionals and scientists.

vi
EBMT and EHA want to express their gratitude to the enormous effort of all
authors in planning and writing the different chapters and especially Isabel Sánchez-
Ortega and Francesco Cerisoli for their continuous and tireless support.
On behalf of EBMT and EHA, we hope this CAR-T cell handbook will be help-
ful in your daily practice.
Hamburg, Germany NicolausKröger
London, UK JohnGribben
Marseille, France ChristianChabannon
Lille, France IbrahimYakoub-Agha
Würzburg, Germany HermannEinsele
Preface

vii
Part I The Science Behind CAR-T Cells
1 Structure of and Signalling Through Chimeric Antigen Receptor . . . 3
Christian Chabannon and Chiara Bonini
2 Genetic Engineering of Autologous or Allogeneic Immune
Effector Cells . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7
Karim Benabdellah, Simone Thomas, and Hinrich Abken
3 What Defines a Good Tumour Antigen? . . . . . . . . . . . . . . . . . . . . . . . . . 11
Emma C. Morris and J. H. F. (Fred) Falkenburg
4 Tumour Escape from CAR-T Cells . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15
Leo Rasche, Luca Vago, and Tuna Mutis
5 CART Initiatives in Europe . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23
Alvaro Urbano-Ispizua and Michael Hudecek
Part II Manufacturing CAR-T Cells: The Supply Chain
6 Providing the Starting Material to the Manufacturer
of an Approved and Commercially Available Autologous
CAR-T Cell Treatment . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31
Halvard Bonig, Christian Chabannon, and Miquel Lozano
7 Receiving, Handling, Storage, Thawing, Distribution,
and Administration of CAR-T Cells Shipped from the
Manufacturing Facility . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 37
Catherine Rioufol and Christian Wichmann
8 Point-of-Care Production of CAR-T Cells . . . . . . . . . . . . . . . . . . . . . . . 45
Julio Delgado, Claire Roddie, and Michael Schmitt
9 Off-the-Shelf Allogeneic CAR-T Cells or Other Immune
Effector Cells . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 51
Stephane Depil and Waseem Qasim
Contents

viii
Part III Clinical Indications for CAR-T Cells
10 Paediatric Acute Lymphoblastic Leukaemia (ALL) . . . . . . . . . . . . . . . 57
Peter Bader, Franco Locatelli, and Christina Peters
11 Adult Acute Lymphoblastic Leukaemia . . . . . . . . . . . . . . . . . . . . . . . . . 61
Elad Jacoby, Nicola Gökbuget, and Arnon Nagler
12 Diffuse Large B Cell Lymphoma and Primary Mediastinal
Lymphoma . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 67
Bertram Glass and Marie José Kersten
13 Mantle Cell Lymphoma . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 75
Noel Milpied and Martin Dreyling
14 Chronic Lymphocytic Leukaemia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 79
Olivier Tournilhac and Peter Dreger
15 Indolent Lymphomas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 83
Franck Morschhauser and Pier Luigi Zinzani
16 Multiple Myeloma . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 87
Ibrahim Yakoub-Agha and Hermann Einsele
17 Developments in Other Haematological Malignancies:
Other Lymphoid Malignancies . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 91
Paolo Corradini and Lorenz Trümper
18 Myeloid Malignancies . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 97
Christophe Ferrand and Alessandro Rambaldi
19 Developments in Solid Tumours . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 105
Paolo Pedrazzoli and John B. A. G. Haanen
Part IV Clinical Management of Patients Treated with CAR-T Cells
20 Bridging Chemotherapy: Adult Acute Lymphoblastic Leukaemia . . . 111
Nicolas Boissel and Fabio Ciceri
21 Bridging to CAR-T Cells in Children, Adolescents,
and Young Adults with ALL . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 115
André Baruchel
22 Bridging Chemotherapy: Relapsed/Refractory
Aggressive B-Cell Lymphoma . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 119
Catherine Thieblemont and Peter Borchmann
23 Bridging Chemotherapy: Follicular Lymphoma,
Mantle Cell Lymphoma, and CLL . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 123
Nico Gagelmann, John Gribben, and Nicolaus Kröger
Contents

ix
24 Bridging Chemotherapy: Multiple Myeloma . . . . . . . . . . . . . . . . . . . . . 127
Salomon Manier, Artur Jurczyszyn, and David H. Vesole
25 Lymphodepleting Conditioning Regimens . . . . . . . . . . . . . . . . . . . . . . . 131
Mohamad Mohty and Monique C. Minnema
26 Management of Cytokine Release Syndrome (CRS) and HLH . . . . . . 135
Francis Ayuk Ayuketang and Ulrich Jäger
27 Management of Immune Effector Cell- Associated
Neurotoxicity Syndrome (ICANS) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 141
Jeremy H. Rees
28 Management of Hypogammaglobulinaemia and B-Cell Aplasia . . . . . 147
Max Topp and Tobias Feuchtinger
29 Management of Myelotoxicity (Aplasia) and Infectious
Complications . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 151
Marion Subklewe and Reuben Benjamin
30 Management of Other Toxicities . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 157
Hermann Einsele and Ibrahim Yakoub-Agha
31 ICU . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 161
Udo Holtick and Elie Azoulay
32 Post-CAR-T Cell Therapy (Consolidation and Relapse):
Acute Lymphoblastic Leukaemia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 165
Jordan Gauthier
33 Post-CAR-T Cell Therapy (Consolidation and Relapse):
Lymphoma . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 169
Didier Blaise and Sabine Fürst
34 Post-CAR-T Cell Therapy (Consolidation and Relapse):
Multiple Myeloma . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 173
Paula Rodríguez-Otero and Jesús F. San Miguel
35 Immune Monitoring . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 177
Susanna Carolina Berger, Boris Fehse, and Marie-Thérèse Rubio
36 Long-Term Follow-Up and Late Effects . . . . . . . . . . . . . . . . . . . . . . . . . 183
Patrick Hayden, Nico Gagelmann, and John Snowden
Part V Access to CAR-T Cells
37 The Regulatory Framework for CAR-T Cells in Europe:
Current Status and Foreseeable Changes AND Centre
Qualification by Competent Authorities and Manufacturers . . . . . . . . 191
Eoin McGrath and Petr Machalik
Contents

x
38 How Can Accreditation Bodies, Such as JACIE
or FACT, Support Centres in Getting Qualified? . . . . . . . . . . . . . . . . . 199
Riccardo Saccardi and Fermin Sanchez-Guijo
39 Educational Needs for Physicians . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 203
Nicolaus Kröger, John Gribben, and Isabel Sánchez-Ortega
40 Education Needs for Nurses in Adult and Paediatric Units . . . . . . . . . 207
Michelle Kenyon, John Murray, Rose Ellard, and Daphna Hutt
41 Role of Pharmacists . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 215
Margherita Galassi and Maria Estela Moreno-Martínez
42 Educational Needs for Cell Processing Facility Personnel . . . . . . . . . . 219
Boris Calmels
43 GoCART . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 221
Soe R. Terwel, Jürgen Kuball, Martin Dreyling,
and Francesco Cerisoli
44 Patient Referral . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 225
John Snowden and Rafael F. Duarte
45 Treatment Coverage and Reimbursement . . . . . . . . . . . . . . . . . . . . . . . 229
Cornelie Haag
46 The Value of CAR-T-cell Immunotherapy in Cancer . . . . . . . . . . . . . . 231
Mohamed Abou-el-Enein and Jordan Gauthier
47 What do Patients Want? The Importance of Patient-reported
Outcomes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 235
Hélène Schoemans, Natacha Bolaños, and Lorna Warwick
Contents

Part I
The Science Behind CAR-T Cells

3
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_1
C. Chabannon (*)
Institut Paoli-Calmettes, Centre de Lutte Contre le Cancer, Centre d’Investigations Cliniques
en Biothérapie, Université d’Aix-Marseille, Inserm CBT-1409, Marseille, France
e-mail: CHAB[email protected].fr
C. Bonini
Experimental Hematology Unit, Division of Immunology, Transplantation and Infectious
Diseases, Vita-Salute San Raffaele University, IRCCS Ospedale San Raffaele Scientic
Institute, Milan, Italy
e-mail: bonini.chiara@hsr.it
1
Structure ofandSignalling Through
Chimeric Antigen Receptor
ChristianChabannon
andChiaraBonini
Chimeric antigen receptor (CAR) is a synthetic transmembrane protein expressed at
the surface of immune effector cells (IECs) that are reprogrammed either invitro or
invivo (June etal. 2018; June and Sadelain 2018). Techniques for genetic engineering
of autologous or allogeneic IECs are described in the next chapter. The synthetic CAR
incorporates several functional domains. The extracellular domain is composed of a
single chain variable fragment (ScFV) of immunoglobulin and recognizes the
“tumour” antigen. The clinical relevance of the selected tumour antigen—with a view
to minimize “on-target/off-tumour” side effects—is discussed in the third chapter of
this section. Bispecic and trispecic CARs are currently being evaluated in preclini-
cal and early clinical trials (Bielamowicz etal. 2018; Shah etal. 2020). The use of an
immunoglobulin domain as the ligand of the target antigen means that recognition is
not restricted to HLA antigens and that CAR-T cells are universally applicable as
opposed to T cell receptor (TCR) transgenic T cells that recognize antigenic peptides
presented in the context of a dened major histocompatibility complex (MHC), limit-
ing clinical applications to subsets of patients with dened HLA typing. The intracel-
lular domain is composed of the intracellular domain of the zeta chain of the CD3
component of the TCR, which will trigger signalling when the CAR engages the tar-
geted ligand. The transmembrane region links the two extracellular and intracellular
domains through the cell membrane and plays an important role in determining the
conformation and exibility of the CAR and its ability to efciently bind the targeted

4
antigen/epitope. Association of only these three functional domains characterized rst
generation CARs, as described in the original publications (Kuwana et al. 1987;
Eshhar etal. 1993). However, full activation of T cells requires the addition of one
(second generation CARs) or two (third generation CARs) domains from costimula-
tory molecules, such as CD28, 4-1BB/CD137, or OX40/CD134, that provide the T
cell costimulatory signal. Currently approved CAR-T cells are second generation
CAR-T cells; as an illustration, the CAR in tisagenlecleucel contains a 4-1BB domain,
while the CAR in axicabtagene ciloleucel contains a CD28 domain. The nature of the
costimulatory domain inuences the ability of CAR-T cells to expand or persist (limit
T cell exhaustion) invivo after infusion into the patient, although it is unclear how this
translates clinically and affects disease control, occurrence of adverse events, and
overall survival due to the lack of head-to-head comparison between approved prod-
ucts. Finally, fourth generation CAR-T cells have been developed for preclinical proj-
ects. These cells, named armoured CAR cells or T cells redirected for universal
cytokine-mediated killing (TRUCKS), encode not only a CAR (usually with one
costimulatory domain, such as in second generation CARs) but also a cytokine, inter-
leukin, pro-inammatory ligand, or chemokine that will counteract the immune sup-
pressive microenvironment that prevails in most solid tumours (Eshhar etal. 1993;
Chmielewski and Abken 2015).
When the CAR engages its ligand, signalling involves several components of the
naturally occurring TCR.These include molecules such as lymphocyte-specic pro-
tein tyrosine kinase (LCK). Some components of the signalling cascade are actionable
with existing drugs, which offers opportunities for pharmacologic modulation of
CAR activity invivo, such as described with tyrosine kinase inhibitors (Mestermann
etal. 2019; Weber etal. 2019); this represents an appealing alternative to the inclusion
of a suicide gene in the CAR construct (Casucci etal. 2013; Gargett and Brown 2014;
Sakemura etal. 2016). Synthetic biology applied to the CAR-T cell eld led to engi-
neering of combinatorial antigen recognition constructs. The “OR” gate strategy (i.e.,
CD19 and CD22) allows CAR-T cell activation upon recognition of at least 1 of the 2
targeted antigens, thus reducing the risk of cancer immune evasion. The “OR” and
“NOT” gate strategies are designed to improve the safety prole of CAR-T cells, since
tumour cells and healthy cells can be discriminated by CAR-T cells based on the
expression pattern of 2 antigens (Weber etal. 2020).
Key Points
• A chimeric antigen receptor is a synthetic transmembrane molecule
encoded by a DNA sequence that combines domains from immunoglobu-
lins, one chain of the T cell receptor, and typically domains from costimu-
latory molecules involved in T cell activation.
• Currently approved and commercially available CAR-T cells are second
generation CAR-T cells that contain a single costimulatory domain.
• The machinery for cell signalling contains actionable elements, thus offer-
ing opportunities for invivo modulation of CAR-T cell activities and miti-
gation of adverse events.
C. Chabannon and C. Bonini

5
References
Bielamowicz K, Fousek K, Byrd TT, Samaha H, Mukherjee M, Aware N, et al. Trivalent
CAR-T cells overcome interpatient antigenic variability in glioblastoma. Neuro-Oncology.
2018;20(4):506–18.
Casucci M, Nicolis di Robilant B, Falcone L, Camisa B, Norelli M, Genovese P, etal. CD44v6-
targeted T cells mediate potent antitumor effects against acute myeloid leukemia and multiple
myeloma. Blood. 2013;122(20):3461–72.
Chmielewski M, Abken H. TRUCKs: the fourth generation of CARs. Expert Opin Biol Ther.
2015;15(8):1145–54.
Eshhar Z, Waks T, Gross G, Schindler DG.Specic activation and targeting of cytotoxic lym-
phocytes through chimeric single chains consisting of antibody-binding domains and the
gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S
A. 1993;90(2):720–4.
Gargett T, Brown MP.The inducible caspase-9 suicide gene system as a “safety switch” to limit on-
target, off-tumor toxicities of chimeric antigen receptor T cells. Front Pharmacol. 2014;5:235.
June CH, Sadelain M.Chimeric antigen receptor therapy. N Engl J Med. 2018;379(1):64–73.
June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC.CAR-T cell immunotherapy for
human cancer. Science. 2018;359(6382):1361–5.
Kuwana Y, Asakura Y, Utsunomiya N, Nakanishi M, Arata Y, Itoh S, etal. Expression of chime-
ric receptor composed of immunoglobulin-derived V regions and T-cell receptor-derived C
regions. Biochem Biophys Res Commun. 1987;149(3):960–8.
Mestermann K, Giavridis T, Weber J, Rydzek J, Frenz S, Nerreter T, et al. The tyrosine kinase
inhibitor dasatinib acts as a pharmacologic on/off switch for CAR-T cells. Sci Transl Med.
2019;11(499):eaau5907.
Sakemura R, Terakura S, Watanabe K, Julamanee J, Takagi E, Miyao K, etal. A Tet-On inducible
system for controlling CD19-chimeric antigen receptor expression upon drug administration.
Cancer Immunol Res. 2016;4(8):658–68.
Shah NN, Johnson BD, Schneider D, Zhu F, Szabo A, Keever-Taylor CA, etal. Bispecic anti-
CD20, anti-CD19 CAR-T cells for relapsed B cell malignancies: a phase 1 dose escalation and
expansion trial. Nat Med. 2020;26(10):1569–75.
Weber EW, Lynn RC, Sotillo E, Lattin J, Xu P, Mackall CL.Pharmacologic control of CAR-T cell
function using dasatinib. Blood Adv. 2019;3(5):711–7.
Weber EW, Maus MV, Mackall CL. The emerging landscape of immune cell therapies. Cell.
2020;181(1):46–62.
Open Access
This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
1 Structure ofandSignalling Through Chimeric Antigen Receptor

7
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_2
K. Benabdellah
Centre for Genomics and Oncological Research (GENYO), Genomic Medicine Department,
Pzer-University of Granada-Andalusian Regional Government, Granada, Spain
e-mail: [email protected]
S. Thomas
Department of Genetic Immunotherapy, Regensburg Center for Interventional Immunology,
Regensburg, Germany
Department of Internal Medicine III, University Hospital Regensburg, Regensburg, Germany
e-mail: [email protected]urg.de
H. Abken (
*)
Department of Genetic Immunotherapy, Regensburg Center for Interventional Immunology,
Regensburg, Germany
e-mail: hinrich.abken@ukr.de
2
Genetic Engineering ofAutologous
orAllogeneic Immune Effector Cells
KarimBenabdellah, SimoneThomas, andHinrichAbken
Manufacturing immune effector cells (T or NK cells) with CAR-encoding DNA
sequences requires efcient and safe genetic engineering procedures. For this pur-
pose, an appropriate genetic vector is chosen according to numerous factors, includ-
ing the vector genome packaging capacity, cellular tropism, genomic integration,
immune toxicity, and other factors. In clinical trials, genomes integrating viral vec-
tors, in particular vectors based on members of the Retroviridae family, such as
retroviruses and lentiviruses, have been successfully used for more than 20years.
These vectors contain an RNA genome that when transcribed into double-stranded
DNA by reverse transcriptase integrates into the genome of the transduced cell.
Several precautions are taken to ensure the safe use of such integrating vectors.
First, the viral genome is split into three different expression constructs to reduce
the risk of recombination events re-establishing replication-competent viruses.
Second, long terminal repeats (LTRs) with their enhancer/promoter sequences are
deleted, resulting in self-inactivated (SIN) vectors to avoid transactivation of cellu-
lar genes in the vicinity of the viral integration site. Third, the viral envelope is
pseudotyped with heterologous glycoproteins, such as gibbon ape leukaemia virus

8
(GALV) or vesicular stomatitis virus (VSV)-G protein, to restrict the cell tropism
for transduction. The viral vectors have undergone generations of modications and
are classied according to their packaging plasmid. During manufacturing, the use
of transduction enhancers, including cationic polymers, lipids, and peptides, such as
Retronectin or Vectofusin-1, which is a histidine-rich cationic amphipathic short
peptide (Jamali etal. 2019), improves the transduction efciencies.
Retroviral vectors modied with the LTRs of the myeloproliferative sarcoma
virus and an improved 5′ untranslated region, named MP71 retroviral vectors, can
achieve high transduction efciencies in human T cells. While retroviral vectors
require actively dividing cells for integration, lentiviral vectors have the capacity to
transduce nondividing or slowly proliferating cells and are currently increasingly
used for genetic modication of T cells in clinical trials. Cycling T cells can ef-
ciently complete the reverse transcription process of the viral vector, facilitate
nuclear import, and enhance the expression of the transgene. Obtaining high virus
titres and ultimately sufcient transduction frequencies for production of CAR-T
cells on a clinical scale and preserving the T cell phenotype and functional proper-
ties after transduction remain a challenge. Despite vector integration into the host
genome, T cells have a negligible risk of transformation; thus far, no leukaemia has
been observed in T cell-based therapy.
Alternatively, articial virus-like particles (VLPs) pseudotyped with VSV-G can
be used for transfer into haematopoietic cells (Mangeot etal. 2019). DNA packed
into transposon vectors, such as sleeping beauty and piggyBac, are transferred to T
cells via electroporation (Kebriaei etal. 2016). Transposon-based genetic engineer-
ing does not require time-consuming and cost-intensive virus production and is
increasingly considered for clinical manufacture of CAR-T cells.
In contrast to integrating DNA transfer technologies, mRNA transfer via electro-
poration or cationic lipid-mediated transfection produces T cells with transient
CAR expression for a few days (Miliotou and Papadopoulou 2020). Such transient
CAR-T cell approaches have been investigated and found to produce antitumour
reactivity for a limited time to avoid any undesirable effects in patients; however,
very few clinical trials using RNA-modied CAR-T cells have been registered.
Genome editing is an upcoming tool to engineer CAR-T cells using specic
endonucleases, including meganucleases (MGNs), transcription activator-like
effector nucleases (TALENs), megaTAL nucleases, zinc-nger nucleases
(ZFNs) and, more recently, clustered regularly interspaced short palindromic
repeat (CRISPR)-Cas9-associated nucleases (Pavlovic etal. 2020). These tech-
nologies allow insertion of a specic DNA sequence at a predened emplace-
ment, such as endogenous genetic locus. While efciently applied in
haematopoietic or mesenchymal stem cell modication for years, genome edit-
ing in primary T cells has only recently been successfully applied towards ef-
cient CAR-T cell engineering. Examples for potential clinical application are
targeting the respective genes for programmed cell death-1 (PD1, CD279), T
cell receptor (TCR) α and β chains, CD52, human leukocyte antigens (HLAs),
and β2-microglobulin (β2M).
K. Benabdellah et al.

9
One major application of genome editing is creating “off-the-shelf” alloge-
neic CAR-T cells to avoid certain limitations associated with autologous T cells,
such as the personalized production process, the several weeks of time required
for manufacturing, and the risk of manufacturing failure. Such allogeneic CAR-T
cells were engineered by genetically eliminating the TCRα constant (TRAC)
locus and/or HLA from the T cell surface, reducing the risk of graft versus host
disease (GvHD) and allograft rejection. In particular, Torikai et al. combined
sleeping beauty transposon- based gene transfer with ZFN-mediated deletion of
TCR α and β chains (Torikai etal. 2012); subsequent approaches also eliminated
the endogenous TCR (Roth etal. 2018; Legut etal. 2018; Osborn etal. 2016).
TALEN-mediated TRAC/CD52 knockout of CD19-specic CAR-T cells
(UCART19) was administered to two patients with relapsed ALL in a proof-of-
concept study, and no GvHD was reported (Qasim etal. 2017). Several approaches
using ZFNs and CRISPR/Cas9, including base editing variants, were used to
eliminate HLA class I expression by targeting β2M (Webber et al. 2019) and
eliminating the HLA class II transactivator CIITA (Kagoya et al. 2020), all
reducing the risk of allogeneic CAR-T cell rejection. To reduce GvHD and frat-
ricide, the CD7 locus was disrupted along with TCRα editing (Gomes-Silva etal.
2017). Eliminating the gene for the TGF-β receptor or PD-1 enhanced CAR-T
cell antitumour potency by reducing repression by the tumour stroma (Tang
etal. 2020).
Genome editing has also been used to insert the CAR-encoding DNA sequence
into the TCR α locus (Eyquem etal. 2017), thereby utilizing the TCR expression
machinery for properly regulated CAR expression. Similarly, CAR-encoding DNA
was inserted into the TCR α locus, and IL-12-encoding DNA was inserted into the
IL2Rα or PDCD1 locus, resulting in CAR-redirected T cell activation along with
IL-12 secretion (Sachdeva etal. 2019). Such genome editing approaches can be
applied to target other signalling pathways to engineer CAR-T cells with therapeu-
tic outputs in a highly regulated manner. Currently, most of these editing technolo-
gies are being explored in mouse models or in a very limited number of patients,
making it difcult to draw a denitive conclusion concerning safety and efcacy in
the long term.
Key Points
• Lentiviral gene transfer is the most frequently applied procedure to engi-
neer CAR-T cells for clinical use.
• Nonviral transposon-mediated DNA transfer is an upcoming technology to
obtain CAR-T cells.
• Allogeneic “off-the-shelf” CAR-T cells are engineered by genetically
eliminating the TCRα constant (TRAC) locus and/or HLA from the T cell
surface, reducing the risk of graft versus host disease (GvHD) and allograft
rejection.
2 Genetic Engineering ofAutologous orAllogeneic Immune Eector Cells

10
References
Eyquem J, Mansilla-Soto J, Giavridis T, etal. Targeting a CAR-T o the TRAC locus with CRISPR/
Cas9 enhances tumour rejection. Nature. 2017;543:113–7.
Gomes-Silva D, Srinivasan M, Sharma S, etal. CD7-edited T cells expressing a CD7-specic CAR
for the therapy of T-cell malignancies. Blood. 2017;130:285–96.
Jamali A, Kapitza L, Schaser T, etal. Highly efcient and selective CAR-gene transfer using CD4-
and CD8-targeted lentiviral vectors. Mol Ther Methods Clin Dev. 2019;13:371–9.
Kagoya Y, Guo T, Yeung B, etal. Genetic ablation of HLA class I, class II, and the T-cell recep-
tor enables allogeneic T cells to be used for adoptive T-cell therapy. Cancer Immunol Res.
2020;8:926–36.
Kebriaei P, Singh H, Huls MH, etal. Phase I trials using sleeping beauty to generate CD19-specic
CAR-T cells. J Clin Invest. 2016;126:3363–76.
Legut M, Dolton G, Mian AA, etal. CRISPR-mediated TCR replacement generates superior anti-
cancer transgenic T cells. Blood. 2018;131:311–22.
Mangeot PE, Risson V, Fusil F, etal. Genome editing in primary cells and invivo using viral-
derived nanoblades loaded with Cas9-sgRNA ribonucleoproteins. Nat Commun. 2019;10:45.
Miliotou AN, Papadopoulou LC.In vitro-transcribed (IVT)-mRNA CAR-T therapy development.
Methods Mol Biol. 2020;2086:87–117.
Osborn MJ, Webber BR, Knipping F, etal. Evaluation of TCR gene editing achieved by TALENs,
CRISPR/Cas9, and megaTAL nucleases. Mol Ther. 2016;24:570–81.
Pavlovic K, Tristán-Manzano M, Maldonado-Pérez N, etal. Using gene editing approaches to ne-
tune the immune system. Front Immunol. 2020;11:570672.
Qasim W, Zhan H, Samarasinghe S, etal. Molecular remission of infant B-ALL after infusion of
universal TALEN gene-edited CAR-T cells. Sci Transl Med. 2017;9:eaaj2013.
Roth TL, Puig-Saus C, Yu R, etal. Reprogramming human T cell function and specicity with
non-viral genome targeting. Nature. 2018;559:405–9.
Sachdeva M, Busser BW, Temburni S, etal. Repurposing endogenous immune pathways to tailor
and control chimeric antigen receptor T cell functionality. Nat Commun. 2019;10:5100.
Tang N, Cheng C, Zhang X, etal. TGF-beta inhibition via CRISPR promotes the long-term ef-
cacy of CAR-T cells against solid tumors. JCI Insight. 2020;5:e133977.
Torikai H, Reik A, Liu PQ, etal. A foundation for universal T-cell based immunotherapy: T cells
engineered to express a CD19-specic chimeric-antigen-receptor and eliminate expression of
endogenous TCR.Blood. 2012;119:5697–705.
Webber BR, Lonetree CL, Kluesner MG, etal. Highly efcient multiplex human T cell engineer-
ing without double-strand breaks using Cas9 base editors. Nat Commun. 2019;10:5222.
Open Access
This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
K. Benabdellah et al.

11
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_3
E. C. Morris (*)
Institute of Immunity and Transplantation, University College London, London, UK
e-mail: [email protected]
J. H. F. (Fred) Falkenburg
Leiden University Medical Center, Leiden, The Netherlands
e-mail: j.h.falkenb[email protected]
3
What Defines aGood Tumour Antigen?
EmmaC.Morris andJ.H.F.(Fred)Falkenburg
Compared to standard anticancer therapies, such as chemotherapy, small molecule
inhibitors and radiation, T cell immunotherapies have the advantages of a high
degree of specicity and durability of response typically associated with cellular
therapies. The functional specicity of a T cell is determined by its antigen recogni-
tion receptor and the target antigen (Bjorkman etal. 1987; Garcia etal. 1996).
The majority of CAR-T cells currently applied in clinical practice do not recog-
nize tumour-specic target antigens but pan-B cell antigens (CD19, CD20, CD22)
or maturation antigens (e.g., BCMA), which are abundantly expressed cell surface
molecules on both malignant and normal cells (Sadelain et al. 2017; June and
Sadelain 2018). In reality, these are only ‘ideal’ or ‘good’ tumour antigens because
the depletion of normal B cells is generally well tolerated. In contrast to endogenous
T cell receptors (TCRs), which are HLA-restricted and recognize peptide-MHC
complexes on the target cell surface, CARs recognize extracellular, membrane-
bound targets. These are typically nonpolymorphic proteins or glycoproteins. This
is advantageous over TCR-mediated recognition because CAR-T cell therapies are
not limited by patient HLA type.
What denes a ‘good’ tumour antigen for recognition by a CAR-T cell?
1. Extracellular expression (i.e., expressed on the cell surface and readily accessible)
2. Uniform or consistent expression on all malignant cells
3. Not subject to downregulation or deletion (i.e., no escape variants). This only
occurs if the antigen is a molecule critical for maintenance of the malignant
population

12
4. Should be expressed on malignant stem cells, and
5. Should not be expressed on normal tissue cells, at least not in nonessential nor-
mal tissues (i.e., tumour specic).
Tumour-Specific Antigens (TSAs)
TSAs are highly specic and typically result from genetic mutations within malig-
nant cells that give rise to neoantigens not present in untransformed (nonmalignant)
cells (Schumacher and Schreiber 2015; Schumacher et al. 2019). By denition,
there is a low likelihood of ‘on-target off-tumour’ toxicity because the tumour anti-
gen is not expressed on normal cells. ‘On-target on-tumour’ toxicities and ‘off-
target off-tumour’ toxicities may occur as a result of CRS or receptor cross-reactivity.
Unfortunately, no nonpolymorphic tumour-specic extracellular target antigens are
known. The only highly specic extracellular tumour target antigens are neopep-
tides presented in the context of (polymorphic) HLA molecules.
Multiple Tumour Antigens Resulting
ina‘Tumour-Specific Phenotype’
Recent studies have demonstrated that simultaneous targeting of two or more target
antigens may improve tumour specicity and reduce the risk of antigen escape
(Shah etal. 2020; Dai etal. 2020). In such cases, one target antigen may be lineage-
specic but not tumour specic, but the combination may be tumour specic. For
CAR-T cells to be fully activated, the target cell must express both target antigens
(i.e., combined antigen expression). This approach is not expected to ameliorate the
risk of CRS, and it is difcult to estimate the risk of ‘on-target off-tumour’ toxicity,
which will depend on the ability of CAR-T cells to discriminate between cells with
combined or single antigen expression. In this case, there would be a potential risk
of ‘off-target off-tumour’ toxicity for single antigens (expression of a single antigen
in normal cells or aberrant antigen expression in normal cells).
Lineage-Specific andDifferentiation Antigens
These antigens are commonly targeted by CAR-T cells and include CD19, CD20,
CD22, and BCMA, which are B cell lineage antigens. Lineage-specic antigens can
be optimal targets in the case of tumours associated with cell lineages and/or tissues
that are nonredundant or temporarily replaceable, such as the B cell lineage, plasma
cells, and thyroid, prostate, and ovarian cells. In these circumstances, their function
can be rescued by a second therapeutic intervention. For example, profound B cell
lymphopenia following CD19 CAR-T cell therapy can result in hypogammaglobu-
linaemia and absent or impaired vaccine responses, requiring long-term immuno-
globulin replacement therapy. More recent developments aimed at generating
E. C. Morris and J. H. F. (Fred) Falkenburg

13
CAR-T cells for treatment of AML and other myeloid malignancies target lineage-
specic and differentiation antigens (i.e., CD33 and CD123) but risk profound cyto-
penia or bone marrow aplasia and depend on the ability to subsequently replace
haematopoietic stem cells and myeloid precursors (Gill etal. 2014). Recent pre-
clinical studies have attempted to ne-tune CAR-T cell responses through the incor-
poration of safety switch mechanisms (Loff etal. 2020). In the case of lineage-specic
antigens, ‘on-target off-tumour’ toxicity is common, resulting in depletion of spe-
cic cell lineages or other cells in the case of aberrant antigen expression. CRS may
be common, due in part to wide antigen expression in both normal and malig-
nant cells.
Lineage-Specific Polymorphic/Heterogeneic Antigens
These target antigens are similar to lineage-specic antigens (above) with the
advantage that only part of the system is eliminated (following CAR-T cell target-
ing) due to intrinsic heterogeneity or antigen expression. Examples include target-
ing immunoglobulin subclasses or kappa versus lambda light chains in association
with immunoglobulin receptors.
References
Bjorkman PJ, Saper MA, Samraoui B, et al. Structure of the human class I histocompatibility
antigen, HLA-A2. Nature. 1987;329(6139):506–12.
Dai H, Wu Z, Jia H, etal. Bispecic CAR-T cells targeting both CD19 and CD22 for therapy
of adults with relapsed or refractory B cell acute lymphoblastic leukemia. J Hematol Oncol.
2020;13(1):30.
Garcia KC, Degano M, Staneld RL, etal. An alphabeta T cell receptor structure at 2.5 A and its
orientation in the TCR-MHC complex. Science. 1996;274(5285):209–19.
Gill S, Tasian SK, Ruella M, etal. Preclinical targeting of human acute myeloid leukemia and
myeloablation using chimeric antigen receptor-modied T cells. Blood. 2014;123:2343–54.
June CH, Sadelain M.Chimeric antigen receptor therapy. N Engl J Med. 2018;379:64–73.
Loff S, Dietrich J, Meyer JE, etal. Rapidly switchable universal CAR-T cells for treatment of
CD123-positive leukemia. Mol Ther Oncolytics. 2020;17:408–20.
Sadelain M, Rivière I, Riddell S.Therapeutic T cell engineering. Nature. 2017;545:423–31.
Key Points
1. Most CAR-T cells and all currently approved products target lineage-
specic antigens.
2. This results in loss of nonmalignant cells that also express these antigens
(e.g., normal B cells).
3. With commercially available CAR-T products, these side effects are man-
ageable but may be more limiting with other novel targets under
development.
3 What Denes aGood Tumour Antigen?

14
Schumacher TN, Schreiber RD.Neoantigens in cancer immunotherapy. Science. 2015;348:69–74.
Schumacher TN, Scheper W, Kvistborg P.Annu Rev Immunol. 2019;37:172–200.
Shah NN, Johnson BD, Schneider D, et al. Bispecic anti-CD20, anti-CD19 CAR-T cells
for relapsed B cell malignancies: a phase 1 dose escalation and expansion trial. Nat Med.
2020;26(10):1569–75.
Open Access
This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
E. C. Morris and J. H. F. (Fred) Falkenburg

15
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_4
L. Rasche
Department of Internal Medicine 2, University Hospital of Würzburg, Würzburg, Germany
Mildred Scheel Early Career Center, University Hospital of Würzburg, Würzburg, Germany
e-mail: rasche_l@ukw.de
L. Vago
Vita-Salute San Raffaele University, Milan, Italy
Unit of Immunogenetics, Leukemia Genomics and Immunobiology, IRCCS San Raffaele
Scientic Institute, Milan, Italy
e-mail: vago.luca@hsr.it
T. Mutis (
*)
Department of Hematology, Amsterdam University Medical Centers, Location VUmc,
Amsterdam, The Netherlands
e-mail: [email protected]
4
Tumour Escape fromCAR-T Cells
LeoRasche, LucaVago, andTunaMutis
Over the past decade, CAR-T cells have emerged as one of the most powerful cel-
lular immune therapy approaches in the battle against haematological malignancies.
Nonetheless, similar to other immunotherapeutic approaches, tumour cells develop
strategies to evade CAR-T cell therapy, often with the support of a highly immuno-
suppressive and protective tumour microenvironment. To date, antigen loss, immune
dysfunction, exhaustion and (microenvironment-mediated) upregulation of anti-
apoptotic pathways have been identied as major modes of tumour escape from
CAR-T cell therapy. This chapter will focus on our current understanding of these
modes of immune escape from CAR-T cells.
Leo Rasche, Luca Vago and Tuna Mutis contributed equally with all other contributors.

16
Immune Escape andCAR-T Cell Resistance Related
toAntigen Loss
Antigen loss represents the ultimate adaptation of a cancer cell to the selective pres-
sure of targeted immunotherapy. While antigen downregulation or dim expression is
a well-known event in lymphoma and myeloma treated with therapeutic IgG anti-
bodies (Plesner etal. 2020; Jilani etal. 2003), complete target loss is a phenomenon
typically occurring after T-cell-based therapy, such as CAR-T cell or T cell engag-
ing bispecic antibodies (TCE) therapy, and rarely after treatment with antibody-
drug conjugates (ADCs).
In B cell malignancies, CD19 loss has been noted in up to 40% of patients with
B cell acute lymphoblastic leukaemia treated with different CAR 19 products
(Orlando etal. 2018). Point mutations in CD19 have been described to lead to
nonfunctional anchoring of the CD19 protein to the cell membrane and conse-
quently to a loss of surface antigen (Orlando etal. 2018). Deleterious mutations
and alternatively spliced CD19 mRNA variants were identied in two other stud-
ies (Asnani etal. 2020; Sotillo etal. 2015). In B-ALL with rearrangement of the
mixed lineage leukaemia (MLL) gene, some patients relapsed with clonally
related acute myeloid leukaemia after treatment with CD19 CAR-T cells, adding
a switch to a CD19- negative myeloid phenotype as another mechanism of resis-
tance (Gardner etal. 2016). In DLBCL, the frequency of CD19 loss after CAR19
axicabtagene ciloleucel (axi-cel) treatment was 33% (Neelapu etal. 2017; Neelapu
etal. 2019), and alternatively spliced CD19 mRNA species could be identied. In
follicular lymphoma and DLBCL treated with CD20 X CD3 bispecic TCE,
CD20 loss relapses were seen, but the frequency is yet to be reported (Bannerji
etal. 2018). Furthermore, a single case of CD22 loss was described after ADC
inotuzumab-ozogamicin treatment in a paediatric patient with B-ALL (Paul etal.
2019). Taken together, antigen loss is a key mechanism of resistance to novel
immunotherapies targeting CD19, CD20, and CD22. In myeloma, downregula-
tion of BCMA was recorded in a signicant proportion of patients following
BCMA CAR-T therapy, but intensity increased back towards baseline in almost
all patients (Cohen etal. 2019). However, three case reports described irreversible
BCMA loss after anti-BCMA CAR-T cell treatment (Da Via etal. 2021; Samur
etal. 2020; Leblay etal. 2020). In two of these cases, homozygous BCMA gene
deletions were identied as the biological underpinning of antigen loss. In the
third case, the authors found a heterozygous BCMA deletion together with a
BCMA mutation, leading to antigen loss. In summary, biallelic events impacting
the BCMA locus represent one molecular mechanism of antigen loss after BCMA
CAR-T therapy. However, these events seem to be rare. In the KarMMa trial, only
4% of patients relapsed without an increase in soluble BCMA, which is thought
to be a biomarker of this type of resistance (Munshi etal. 2021). Heterozygous
BCMA deletions, present in approximately 7% of anti-BCMA naïve patients, rep-
resent a risk factor for BCMA loss-relapse after T-cell-based therapy (Da Via
etal. 2021). While a plethora of alternative antigens, such as FCRH5 or GPRC5D,
are currently being investigated in early clinical trials (Rasche et al. 2020),
L. Rasche et al.

17
antigen loss for these targets has not been reported thus far. However, MM is a
disease associated with high frequencies of copy number aberrations, including
deletions impacting genes encoding immunotherapy targets, and we expect bial-
lelic events leading to antigen loss to also be relevant for MM targets other than
BCMA. Multispecic CAR-T cells or combinations of monospecic targeted
immunotherapies may overcome antigen loss in future trials (Fernández de Larrea
etal. 2020).
Immune Dysfunction andExhaustion ofCAR-T Cells
In addition to antigen loss, a number of other mechanisms also limit or abrogate the
effective recognition of cancer cells by CAR-T cells, either directly conveyed by
tumour cells or through rewiring of the microenvironment. In preclinical models,
especially in solid tumours, it was shown that tumour-inltrating CAR-T cells
undergo rapid loss of functionality, limiting their therapeutic efcacy. This hypore-
sponsiveness appears to be reversible when the T cells are isolated away from the
tumour and is associated with upregulation of intrinsic T cell inhibitory enzymes
(diacylglycerol kinase and SHP-1) and with the expression of surface inhibitory
receptors (PD1, LAG3, TIM3, and 2B4) (Moon etal. 2014).
Additionally, in patients with diffuse large B cell lymphoma (DLBCL) treated
with axicabtagene ciloleucel (axi-cel), it has been shown that tumour-inltrating
CAR-T cells express the inhibitory receptor PD1 and that they represent only a
minor fraction of the immune cells detectable in the tumour (Chen etal. 2020).
Of note, immunogenic chemotherapy can enhance the recruitment of CAR-T
cells to the tumour bed by inducing the release of chemokines from monocytes,
and this can potently synergize with immune checkpoint blockade (Srivastava
et al. 2021). In another recent study in DLCBL, interferon (IFN) signalling
expression, along with high blood levels of monocytic myeloid-derived suppres-
sor cells (M-MDSCs), IL-6 and ferritin, was associated with a lack of a durable
response to axi-cel. The authors showed that high IFN signalling is associated
with the expression of multiple checkpoint ligands, including PD-L1, on lym-
phoma cells and that these levels were higher in patients who lacked a durable
response to CAR-T therapy (Jain etal. 2021). However, impairment of IFN sig-
nalling, such as through mutations or downmodulation of JAK2 and other path-
way components, can confer tumour cell resistance to killing by CAR-redirected
T cells (Arenas etal. 2021).
These ndings have direct implications for the design of next-generation CAR-T
cell protocols: a number of strategies are now being explored to combine immune
checkpoint blockade with CAR-T cell therapy, either by coinfusion of genetically
modied lymphocytes with monoclonal antibodies or by engineering the cell to
produce the relevant scFv (Carneiro and El-Deiry 2020), be resistant to inhibitory
signals (Cullen et al. 2010), or even transform signals under activating stimuli
(Sutton etal. 2000). Moreover, novel promising compounds have been shown to
counteract the activity of T cell inhibitory enzymes (Moon etal. 2014).
4 Tumour Escape fromCAR-T Cells

18
Microenvironment-Mediated Tumour Resistance toCAR-T Cells
Immune suppression or exhaustion is not the only mechanism by which tumour
cells can become less susceptible to CAR-T cell-mediated cytotoxicity. In many
haematological cancers, the bone marrow tumour microenvironment (BMME) is
known to upregulate antiapoptotic mechanisms in tumour cells through tight cross-
talk of mesenchymal stromal cells (MSCs) and tumour cells. Remarkably, tumour
cell lysis by T and NK cells is also largely mediated via activation of extrinsic and
intrinsic apoptosis pathways (Hanabuchi et al. 1994; Falschlehner et al. 2009;
Carneiro and El-Deiry 2020; Cullen etal. 2010; Sutton etal. 2000). Thus, the idea
that BMMSCs might also induce resistance to T and MK cell-mediated cytotoxic
activity through upregulation of antiapoptotic mechanisms has recently been tested,
and the results showed that MM cell-BMMSC interactions can indeed protect MM
cells from conventional cytotoxic T cells and from (daratumumab redirected) NK
cells (McMillin etal. 2012; de Haart etal. 2013; de Haart etal. 2016). These studies
were recently extended to CAR-T cells by testing a panel of nine different
MM-reactive CAR-T cells that were reactive to three different MM-associated anti-
gens (CD138, BCMA, and CD38) with different target afnities and with different
costimulatory domains (CD28, 4-1BB, or CD28 plus 4-1BB) (Holthof etal. 2021a).
In the absence of BMMSCs, BCMA
bb2121
CAR-T cells, high afnity CD38 CAR-T
cells, and intermediate afnity CD38 CAR-T cells containing CD28 costimulatory
domains showed high levels of anti-MM cell lysis, whereas other CAR-T cells
showed moderate cytotoxic activity against MM cells. BMMSCs did not modulate
the lytic activity of highly lytic CAR-T cells but readily protected MM cells against
all other CAR-T cells with intermediate killing capacity. Overall, a strong inverse
correlation was demonstrated between the lytic capacity of the CAR-T cells and the
extent of BMMSC-mediated protection. Furthermore, the BMMSC-mediated pro-
tection of MM cells from these CAR-T cells was readily abrogated by inhibition of
survivin, MCL-1, and Xiap using the small molecule FL118. Thus, the results con-
rmed that BMMSC-mediated immune resistance was mediated by negative regula-
tion of apoptotic pathways. In addition, the importance of the tumour stroma in the
efcacy of CAR-T cells has also been suggested in a solid tumour mouse model,
where destruction of the tumour stroma contributed to eradication of large tumours
by HER2-specic CAR-T cells (Textor etal. 2014). Based on these studies, over-
coming BMMSC-mediated immune resistance seems possible by increasing the
overall avidity and killing activity of CAR-T cells. This may be achieved by design-
ing CARs containing high afnity antigen recognition domains, tandem CARs, or
dual CAR strategies (van der Schans etal. 2020). Alternatively, using the CD28
costimulatory domain (Drent etal. 2019; Drent etal. 2017) or engineering CAR-T
cells with cytotoxic effector molecules can upregulate CAR-T cell activity. Indeed,
it has recently been demonstrated that BMSMSC-mediated immune resistance
towards the NK cell line KHYG-1 can be abrogated by engineering it with a CD38
CAR and/or with a DR5-specic, optimized TRAIL variant (Holthof etal. 2021b).
CAR-T cells may also be equipped with caspase-independent apoptotic molecules,
such as granzyme-A (Borner and Monney 1999).
L. Rasche et al.

19
In addition, a number of earlier and recent studies indicate the importance of
apoptotic pathways for the efcacy of other CAR-T cells. For instance, CD19
CAR-T cells were previously found to benet from combination with the BCL-2
inhibitor ABT-737 (Karlsson etal. 2013). Recently, similar results were observed
when third-generation CD19 CAR-T cells were combined with another BCL-2
inhibitor, ABT199 (Yang etal. 2019). Finally, two independent loss-of-function
screens in ALL cell lines identied impaired death receptor pathways as another
mechanism of resistance to CD19-targeted CAR therapy. Loss of FADD, BID, and
tumour necrosis factor-related apoptosis-inducing ligand 2 (TRAIL2) in leukaemia
cells was shown to render them more resistant to cytotoxicity and to drive T cell
exhaustion upon prolonged stimulation (Singh etal. 2020; Dufva etal. 2020). The
combination of CAR-T cells with the SMAC mimetic compound birinapant sig-
nicantly improved the lysis of malignant cells (Dufva etal. 2020). Thus, when
increasing the lytic capacity of CAR-T cells is not possible or desirable, especially
if the target antigen is not entirely tumour-specic, tumour cells can be made more
sensitive by combining CAR-T cells with small molecules targeting regulatory
proteins in the intrinsic and extrinsic apoptotic pathways, as shown in these studies.
References
Arenas EJ, Martínez-Sabadell A, Rius Ruiz I, Román Alonso M, Escorihuela M, Luque A, etal.
Acquired cancer cell resistance to T cell bispecic antibodies and CAR-T targeting HER2
through JAK2 down-modulation. Nature. Communications. 2021;12(1):1237.
Asnani M, Hayer KE, Naqvi AS, Zheng S, Yang SY, Oldridge D, etal. Retention of CD19 intron 2
contributes to CART-19 resistance in leukemias with subclonal frameshift mutations in CD19.
Leukemia. 2020;34:1202–7.
Bannerji R, Arnason JE, Advani R, Brown JR, Allan JN, Ansell SM, etal. Emerging clinical activ-
ity of REGN1979, an anti-CD20 x anti-CD3 bispecic antibody, in patients with relapsed/
refractory follicular lymphoma (FL), diffuse large B-cell lymphoma (DLBCL), and other
B-cell non-Hodgkin lymphoma (B-NHL) subtypes. Blood. 2018;132(Suppl 1):1690.
Borner C, Monney L.Apoptosis without caspases: an inefcient molecular guillotine? Cell Death
Differ. 1999;6(6):497–507.
Carneiro BA, El-Deiry WS. Targeting apoptosis in cancer therapy. Nat Rev Clin Oncol.
2020;17(7):395–417.
Chen P-H, Lipschitz M, Weirather JL, Jacobson C, Armand P, Wright K, etal. Activation of CAR
and non-CAR-T cells within the tumor microenvironment following CAR-T cell therapy. JCI
Insight. 2020;5(12):e134612.
Key Points
1. Loss of expression of the target antigen on tumour cells via the selective
immune pressure of CAR-T cells is a major mechanism of CAR-T cell
therapy failure.
2. T cell exhaustion of CAR-T cells can decrease their function.
3. Multiple other cells in the tumour microenvironment can contribute to
inhibition of CAR-T cell function.
4 Tumour Escape fromCAR-T Cells

20
Cohen AD, Garfall AL, Stadtmauer EA, Melenhorst JJ, Lacey SF, Lancaster E, etal. B cell matu-
ration antigen-specic CAR-T cells are clinically active in multiple myeloma. J Clin Invest.
2019;129(6):2210–21.
Cullen SP, Brunet M, Martin SJ. Granzymes in cancer and immunity. Cell Death Differ.
2010;17(4):616–23.
Da Via MC, Dietrich O, Truger M, Arampatzi P, Duell J, Heidemeier A, etal. Homozygous BCMA
gene deletion in response to anti-BCMA CAR-T cells in a patient with multiple myeloma. Nat
Med. 2021;27:616–9.
Drent E, Themeli M, Poels R, de Jong-Korlaar R, Yuan H, de Bruijn J, etal. A rational strategy for
reducing on-target off-tumor effects of CD38-chimeric antigen receptors by afnity optimiza-
tion. Mol Ther. 2017;25(8):1946–58.
Drent E, Poels R, Ruiter R, van de Donk N, Zweegman S, Yuan H, etal. Combined CD28 and
4-1BB costimulation potentiates afnity-tuned chimeric antigen receptor-engineered T cells.
Clin Cancer Res. 2019;25(13):4014–25.
Dufva O, Koski J, Maliniemi P, Ianevski A, Klievink J, Leitner J, etal. Integrated drug prol-
ing and CRISPR screening identify essential pathways for CAR-T cell cytotoxicity. Blood.
2020;135(9):597–609.
Falschlehner C, Schaefer U, Walczak H. Following TRAIL’s path in the immune system.
Immunology. 2009;127(2):145–54.
Fernández de Larrea C, Staehr M, Lopez AV, Ng KY, Chen Y, Godfrey WD, etal. Dening an
optimal dual-targeted CAR-T cell therapy approach simultaneously targeting BCMA and
GPRC5D to prevent BCMA escape–driven relapse in multiple myeloma. Blood Cancer
Discovery. 2020;1(2):146–54.
Gardner R, Wu D, Cherian S, Fang M, Hana LA, Finney O, etal. Acquisition of a CD19-negative
myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T cell
therapy. Blood. 2016;127(20):2406–10.
de Haart SJ, van de Donk NW, Minnema MC, Huang JH, Aarts-Riemens T, Bovenschen N, etal.
Accessory cells of the microenvironment protect multiple myeloma from T-cell cytotoxicity
through cell adhesion-mediated immune resistance. Clin Cancer Res. 2013;19(20):5591–601.
de Haart SJ, Holthof L, Noort WA, Minnema MC, Emmelot ME, Aarts-Riemens T, et al.
Sepantronium bromide (YM155) improves daratumumab-mediated cellular lysis of multiple
myeloma cells by abrogation of bone marrow stromal cell-induced resistance. Haematologica.
2016;101(8):e339–42.
Hanabuchi S, Koyanagi M, Kawasaki A, Shinohara N, Matsuzawa A, Nishimura Y, et al. Fas
and its ligand in a general mechanism of T-cell-mediated cytotoxicity. Proc Natl Acad Sci.
1994;91(11):4930–4.
Holthof L, van der Schans J, Katsarau A, Poels R, Gelderloos A, van Hal SE, etal. Bone marrow
mesenchymal stromal cells can render multiple myeloma cells resistant to cytotoxic machinery
of CAR-T cells through inhibition of apoptosis. Clin Cancer Res. 2021a;27(13):3793–803.
Holthof L, van der Horst HJ, Gelderloos A, Poels R, Li F, Groen R, etal. Bone marrow mesenchy-
mal stromal cell-mediated resistance in multiple myeloma against NK cells can be overcome
by introduction of CD38-CAR or TRAIL-variant. Hema. 2021b;5(5):e561.
Jain MD, Zhao H, Wang X, Atkins R, Menges M, Reid K, etal. Tumor interferon signaling and
suppressive myeloid cells associate with CAR-T cell failure in large B cell lymphoma. Blood.
2021;137(19):2621–33.
Jilani I, O’Brien S, Manshuri T, Thomas DA, Thomazy VA, Imam M, et al. Transient down-
modulation of CD20 by rituximab in patients with chronic lymphocytic leukemia. Blood.
2003;102(10):3514–20.
Karlsson SCH, Lindqvist AC, Fransson M, Paul-Wetterberg G, Nilsson B, Essand M, et al.
Combining CAR-T cells and the Bcl-2 family apoptosis inhibitor ABT-737 for treating B-cell
malignancy. Cancer Gene Ther. 2013;20(7):386–93.
Leblay N, Maity R, Barakat E, McCulloch S, Duggan P, Jimenez-Zepeda V, etal. Cite-Seq pro-
ling of T cells in multiple myeloma patients undergoing BCMA targeting CAR-T or bites
immunotherapy. Blood. 2020;136(Suppl 1):11–2.
L. Rasche et al.

21
McMillin DW, Delmore J, Negri JM, Vanneman M, Koyama S, Schlossman RL, etal. Compartment-
specic bioluminescence imaging platform for the high-throughput evaluation of antitumor
immune function. Blood. 2012;119(15):e131–8.
Moon EK, Wang LC, Dol DV, Wilson CB, Ranganathan R, Sun J, etal. Multifactorial T-cell
hypofunction that is reversible can limit the efcacy of chimeric antigen receptor-transduced
human T cells in solid tumors. Clin Cancer Res. 2014;20(16):4262–73.
Munshi NC, Anderson LD Jr, Shah N, Madduri D, Berdeja J, Lonial S, etal. Idecabtagene Vicleucel
in relapsed and refractory multiple myeloma. N Engl J Med. 2021;384(8):705–16.
Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, etal. Axicabtagene
ciloleucel CAR-T cell therapy in refractory large B-cell lymphoma. N Engl J Med.
2017;377(26):2531–44.
Neelapu SS, Rossi JM, Jacobson CA, Locke FL, Miklos DB, Reagan PM, etal. CD19-loss with
preservation of other B cell lineage features in patients with large B cell lymphoma who
relapsed post-axi-cel. Blood. 2019;134(Suppl 1):203.
Orlando EJ, Han X, Tribouley C, Wood PA, Leary RJ, Riester M, et al. Genetic mechanisms
of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat Med.
2018;24(10):1504–6.
Paul MR, Wong V, Aristizabal P, Kuo DJ.Treatment of recurrent refractory pediatric pre-B acute
lymphoblastic leukemia using Inotuzumab Ozogamicin monotherapy resulting in CD22
antigen expression loss as a mechanism of therapy resistance. J Pediatr Hematol Oncol.
2019;41(8):e546–e9.
Plesner T, van de Donk N, Richardson PG.Controversy in the use of CD38 antibody for treatment
of myeloma: is high CD38 expression good or bad? Cell. 2020;9(2):378.
Rasche L, Hudecek M, Einsele H.What is the future of immunotherapy in multiple myeloma?
Blood. 2020;136(22):2491–7.
Samur MK, Fulciniti M Aktas-Samur A, Hamid Bazarbachi A, Tai Y, Campbell TB, Petrocca F,
Hege K, Kaiser S, Anderson K, Munshi NC (2020) Biallelic loss of BCMA triggers resistance
to anti-BCMA CAR-T cell therapy in multiple myeloma (abstract #721). 62nd ASH annual
meeting and exposition
van der Schans JJ, van de Donk N, Mutis T. Dual targeting to overcome current challenges in
multiple myeloma CAR-T cell treatment. Front Oncol. 2020;10:1362.
Singh N, Lee YG, Shestova O, Ravikumar P, Hayer KE. Impaired death receptor signaling in
leukemia causes antigen-independent resistance by inducing CAR-T cell dysfunction.
2020;10(4):552–67.
Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, etal. Convergence of acquired
mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy.
Cancer Discov. 2015;5(12):1282–95.
Srivastava S, Furlan SN, Jaeger-Ruckstuhl CA, Sarvothama M, Berger C, Smythe KS, et al.
Immunogenic chemotherapy enhances recruitment of CAR-T cells to lung tumors and
improves antitumor efcacy when combined with checkpoint blockade. Cancer Cell.
2021;39(2):193–208.e10.
Sutton VR, Davis JE, Cancilla M, Johnstone RW, Ruei AA, Sedelies K, etal. Initiation of apopto-
sis by granzyme B requires direct cleavage of bid, but not direct granzyme B-mediated caspase
activation. J Exp Med. 2000;192(10):1403–14.
Textor A, Listopad JJ, Wuhrmann LL, Perez C, Kruschinski A, Chmielewski M, etal. Efcacy of
CAR-T cell therapy in large tumors relies upon stromal targeting by IFNgamma. Cancer Res.
2014;74(23):6796–805.
Yang M, Wang L, Ni M, Schubert M-L, Neuber B, Hückelhoven-Krauss A, et al. The effect
of apoptosis inhibitor blockade agents on the third generation CD19 CAR-T cells. Blood.
2019;134(Suppl 1):5620.
4 Tumour Escape fromCAR-T Cells

22
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
L. Rasche et al.

23
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_5
A. Urbano-Ispizua
Institute of Hematology and Oncology. Hospital Clinic of Barcelona. University of
Barcelona, Barcelona, Spain
e-mail: [email protected]
M. Hudecek (
*)
Department of Internal Medicine II, University Hospital of Würzburg, Würzburg, Germany
e-mail: Hudecek_M@ukw.de
5
CART Initiatives inEurope
AlvaroUrbano-Ispizua andMichaelHudecek
The efcacy of chimeric antigen receptor T cells (CARTs) in B cell neoplasms,
ALL, large B cell lymphoma, and now multiple myeloma has been one of the great
achievements in the ght against cancer in recent decades (Porter et al. 2011).
However, there is still a need to increase the proportion of responses (especially in
NHL) (Locke etal. 2019) and to decrease the proportion of relapse (especially in
ALL) (Grupp etal. 2013). More importantly, currently, commercial CAR-T prod-
ucts are not available for T cell neoplasms, myeloid malignant haemopathies, or
solid tumours. As a reection of the necessary efforts to expand the efcacy of
CARTs, more than 500 clinical trials are currently underway worldwide, mostly led
by American or Chinese groups. Unfortunately, European institutions are under-
represented in these initiatives. It is our duty to push and harmonize European aca-
demic clinical trials. We identied 35 early clinical trials promoted by European
groups in Eudract and ClinTrialsGov (20 February 2021) (Table5.1). Among them,
20 are initiatives from academic institutions, and 15 are initiatives from European
companies allied with European academic institutions. In this summary, CART
clinical trials promoted by European academic centres or by small to medium
European companies are listed. The aim is to inform European groups treating hae-
mato-oncology diseases of the current situation in this eld, facilitating the inclu-
sion of patients in these clinical trials. We aim to support the groups promoting
these studies to increase collaboration.

24
Table 5.1 Ongoing CART clinical trials in Europe
A. Urbano-Ispizua and M. Hudecek

25
(continued)
5 CART Initiatives inEurope

26
Table 5.1 (continued)
A. Urbano-Ispizua and M. Hudecek

27
Twelve European institutions are responsible for the 20 academic clinical trials
(University College London, n= 4; Hospital Clinic of Barcelona, n = 3; Great
Ormond Street Hospital, n=2; Bambino Gesù, n=2; University of Uppsala, n=2;
King’s College, n=1; Matilde Tettamanti, n=1; Hospital Sant Pau, n=1; University
of Heidelberg, n=1; and University of Wurzburg, n=1). The most frequent target
is CD19 (n=12; for ALL, n=5; NHL, n=1; all B-lymphoid neoplasms, n=6).
Other targets are BCMA (n=1; MM), CD30 (n=1; HL and T-NHL), SLAMF7
(n=1; MM), GD2 (n=2, neuroblastoma), ErbBR (n=1, neck and head tumours),
Fap (n=1, mesothelioma), and IL-1 RAP (n=1, CML). Of the 20 clinical trials, 8
only included adults, 5 only included children, and 7 included all ages.
There were 16 additional clinical trials promoted by seven European pharma
companies (Autolous, n=6; Miltenyi, n=3; Servier, n=2; MolMed, n=1; Celyad,
n=1; Cellectis, n=1; TcBiopharm, n=1, BioNTech, n=1). Again, the most fre-
quent target is CD19 (n=4; for ALL, n=2; NHL, n=1; all B-lymphoid neoplasms,
n=1). Other targets are dual CD19/CD20 or CD19/CD22 (n=3; ALL, n=1; NHL,
n=2), CD20 (n=2; melanoma, lymphoma), BCMA (n=1; MM), CD123 (n=1;
AML), CD33 (n=1, AML), NKG2D (n=1, colon cancer), CD44v6 (n=1, MM),
TRCDB1 (n=1, T-NHL), and CLDN6 (n=1; colon cancer).
Hopefully, this list will grow as more clinical trials are set up. We intend to com-
pile an ad hoc workshop to provide more comprehensive data, such as the charac-
teristics of the genetic construct (type of costimulatory molecule, 2nd- or
third-generation CAR), the vector (viral or nonviral), and the method of expansion
(automated or manual) and the plans of these groups to go beyond a particular clini-
cal trial (hospital exemption, EMA). We believe this information will be useful to
increase efforts and fuel this eld in Europe.
Two initiatives have recently been launched to foster collaboration and increase
CART activity in Europe: GoCART and T2 EVOLVE Consortium.
GoCART is a strategic partnership between EBMT and EHA that includes a
multistakeholder coalition of patient representatives, health care professionals,
pharmaceutical companies, regulators, health technology assessment (HTA) bodies,
reimbursement agencies, and medical organizations. Some of its most important
aims include the following:
• Collaborate and share data and knowledge.
• Promote harmonization of data collection, education, standards of care, regula-
tory approval, centre qualication, and reimbursement processes.
• Set up a pre- and postmarketing registry that supports regulatory and shared
research purposes.
• Develop a cellular therapy education and information program for patients and
health care professionals.
T2EVOLVE is an alliance of academic and industry leaders in cancer immuno-
therapy under the European Union’s Innovative Medicines Initiative (Supported
from the European Union’s Horizon 2020 Research and Innovation Programme).
The key objective of T2EVOLVE is to accelerate development and increase the
5 CART Initiatives inEurope

28
awareness and access of cancer patients to immunotherapy with immune cells that
harbour a genetically engineered TCR or CAR.Simultaneously, T2EVOLVE aims
to provide guidance on sustainable integration of these treatments into the EU health
care system. The T2EVOLVE consortium aims to achieve its goal by working on
and improving the state of the art in the following key aspects:
• Selection of optimal lymphodepletion regimens.
• Optimization of preclinical models for the best safety and efcacy prediction.
• To involve and guide patients throughout their clinical journey.
• Denition of gold standard analytical methods pre- and post-engineered T cell
infusion.
• Production of GMP guidance and establishment of standard product proles.
• To produce excellent cancer therapies accessible to all European patients.
References
Grupp SA, Kalos M, Barrett D, etal. Chimeric antigen receptor–modied T cells for acute lym-
phoid leukemia. N Engl J Med. 2013;368:1509–18.
Locke FL, Ghobadi A, Jacobson CA, etal. Long-term safety and activity of axicabtagene ciloleu-
cel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1–2 trial.
Lancet Oncol. 2019;20:31–42. https://doi.org/10.1016/S1470- 2045(18)30864- 7.
Porter DL, Levine BL, Kalos M, etal. Chimeric antigen receptor–modied T cells in chronic lym-
phoid leukemia. N Engl J Med. 2011;365:725–33.
Open Access
This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
Key Points
1. Two initiatives have been launched to foster collaboration and increase
CART activity in Europe: GoCART and T2 EVOLVE Consortium.
2. A large number of CAR-T cell clinical trials are underway.
3. Most clinical trials are occurring in the USA and China, and although
Europe has lagged behind, there is evidence of increasing activity.
4. Initiatives to enhance clinical trial activity and cooperation across Europe
are needed, and various initiatives are planned to facilitate this.
A. Urbano-Ispizua and M. Hudecek

Part II
Manufacturing CAR-T Cells: The Supply Chain

31
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_6
H. Bonig
Translational Development of Cellular Therapeutics, Institute for Transfusion Medicine and
Immunohematology, Goethe University, Frankfurt, Germany
Medicine/Hematology, University of Washington, Seattle, WA, USA
Transfusion Medicine, University of Ljubljana, Ljubljana, Slovenia
e-mail: [email protected]
C. Chabannon (
*)
Institut Paoli-Calmettes, Centre de Lutte Contre le Cancer, Centre d’Investigations Cliniques
en Biothérapie, Université d’Aix-Marseille, Inserm CBT-1409, Marseille, France
e-mail: CHAB[email protected].fr
M. Lozano
Department of Hemotherapy and Hemostasis, Institute of Hematology and Oncology, Clinic
University Hospital, University of Barcelona, IDIBAPS, Barcelona, Catalonia, Spain
e-mail: [email protected]
6
Providing theStarting Material
totheManufacturer ofanApproved
andCommercially Available Autologous
CAR-T Cell Treatment
HalvardBonig, ChristianChabannon
, andMiquelLozano
Introduction
CAR-T cell manufacturing starts from a collection of mononuclear cells (MNCs,
although specically only T lymphocytes will be used for the preparation) from the
patient using apheresis. Although several initiatives are working on the develop-
ment of allogeneic CAR-T cells, currently only CAR-T therapies of autologous
origin are approved in the European Union. The present chapter only discusses
already or soon-to-be marketed autologous CAR-T cells and excludes investiga-
tional CAR-T cells or rare CAR-T cells approved in the context of hospital exemp-
tion, such as the ARI-001 product (Ortiz-Maldonado et al. 2021); on this topic,
please refer to Chap. 3.
Institutions aspiring to be CAR-T centres must generate sufcient apheresis
capacity to ensure immediate access to apheresis slots; apheresis capacity must
grow in synchrony with the CAR-T program (Tables 6.1, 6.2, 6.3, 6.4, and 6.5).

32
Table 6.1 Before apheresis collection
Apheresis units might require a visit during the screening of patients for CAR-T therapies
In the visit the following aspects should be evaluated (Yakoub-Agha etal. 2020):
– Discontinue drugs that affect the number and functionality of circulating T lymphocytes
(e.g., steroids, immune-suppressors, and chemotherapy) as long as possible before
apheresis. Minimal stopping rules are dened in institutional guidelines and manufacturing
authorization holder (MAH) instructions. Steroids are usually stopped at least 3–7days
before apheresis.
– Evaluate for systemic infection, particularly in patients with an intravenous central line.
Bacteremia is a relative contraindication for MNC apheresis due to the risk of
contamination of the product. Note, however, that a contaminated blood product—
although denitely not ideal—is not always rejected by the manufacturer because the
manufacturing process may plan for the addition of antibiotics.
– Assess venous access, ideally including an ultrasound evaluation if upper arm peripheral
veins are not adequate by palpation (Gopalasingam etal. 2017) before deciding that a
central line catheter must be placed. Blood can be collected from most adult patients via
peripheral access sites. Central catheters are most often needed in low-weight children.
Indwelling catheters are suitable (Jarisch etal. 2020).
– Evaluate whether any form of sedation is necessary (mostly for the paediatric population)
through a joint evaluation between the paediatricians and apheresis medical director or
practitioner.
– Review complete blood count (CBC) and differential (absolute lymphocyte count) results
to calculate target process volume.
– Check the MCV to rule out the presence of beta thalassemia minor. Microcytosis leads to
abnormal sedimentation behaviour, necessitating modication of apheresis settings
(Constantinou etal. 2017).
– Note height and weight and calculate total blood volume. In particular, in low-weight
children, consider the need for priming of the apheresis tubing set with irradiated red blood
cell concentrate (which needs to be pre-ordered so as not to delay initiation of apheresis).
– If the patient is transfused with cellular components in the days prior to apheresis
collection, they must be gamma irradiated.
– Note concurrent medications, especially anti-hypertensives. Angiotensin-converting
enzyme inhibitors and beta-blockers increase the risk of hypotension reactions during
apheresis.
– Evaluate electrolyte levels: potassium, (ionized) calcium, and magnesium levels drop
during apheresis and thus can become critically low if already abnormal before apheresis
(Stenzinger and Bonig 2018).
– Negative infectious markers of HIV, HBV, and HCV (in some countries also HTLV-1 and
syphilis) must be available within 30days of collection, in compliance with applicable
laws and regulations for autologous cell products. MAH will require that results be
available upon shipment of the starting material to the manufacturing site.
H. Bonig et al.

33
Table 6.2 Designing apheresis collection
– Select the apheresis platform suitable for the collection. Depending on the platform
selected, the characteristics of the product collected might vary signicantly. For instance,
if an Amicus separator (Fresenius-Kabi, Bad Homburg, Germany) is used, the platelet
content is very low in comparison to Spectra Optia (Terumo BCT, Lakewood, CO, USA)
(Cid etal. 2019), see Table6.3.
– Check if the total blood volume of the patient requires priming of the apheresis separator.
– Check lymphocyte count to dene the blood volume to be processed, see Table6.4 for
apheresis targets of different manufacturers.
– Consider prophylactic administration of calcium and magnesium during the collection
depending on the volume to be processed (Sörensen etal. 2013).
– Evaluate the haemoglobin and platelet count, consider transfusion prior to or after
apheresis depending on circumstances. A haemoglobin level of 7g/dL, better yet 8g/dL,
prior to apheresis facilitates establishment of interphase during apheresis. Spectra Optia
will reduce platelet count by approximately 11% per total blood volume processed.
– Evaluate the potassium level and consider supplementation.
Table 6.3 Characteristics of the apheresis platforms typically used for collection
Amicus Spectra optia
Manufacturer Fresenius-Kabi (Bad
Homburg, Germany)
TerumoBCT, Lakewood, Co, USA
Software kit Continuous MNC
(CMNC)
MNC
Flow type Discontinuous Continuous Dual-stage separation
Flow rate 10–80 (85) mL/min
a
10–142mL/min 10–125mL/min
Operation Automatic Semi-automatic Semi-automatic
Mononuclear cell
collection
Elutriation Aspiration Aspiration followed
by elutriation
Platelets Returned to the donor Collected Partially returned to
donor
a
Depending on the leukocyte count
6 Providing the Starting Material to the Manufacturer of an Approved…

34
Table 6.4 Collection requirements of different manufacturers of CAR-T therapies
Product
Axicabtagene
ciloleucel
Brexucabtagene
autoleucel Tisagenlecleucel
Lisocabtagene
maraleucel
Idecabtagene
vicleucel
Ciltacabtagene
autoleucel
Registered name Yescarta
®
Tecartus
®
Kymriah
®
Breyanzi
®
Abecma
®
Manufacturer Gilead Gilead Novartis Juno-Celgene-
BMS
BlueBird
Bio-Celgene-
BMS
Legend
Therapeutics-
Janssen
Antigen
recognized
CD19 CD19 CD19 CD19 BCMA BCMA
Target cell dose
during apheresis
a
5–10×10
9
MNCs 1–4×10
9
CD3
+
cells
≥2×10
9
TNCs
≥3% CD3+ of TNCs
(rounding rules apply)
Target volume to
be processed
a
12–15L 6–10L
a
Manufacturer’s recommendation
MNCs mononuclear cells, TNCs total nucleated cells
H. Bonig et al.

35
References
Cid J, Carbasse G, Alba C, Perea D, Lozano M.Leukocytapheresis in nonmobilized donors for
cellular therapy protocols: evaluation of factors affecting collection efciency of cells. J Clin
Apher. 2019;34:672–9.
Constantinou VC, Bouinta A, Karponi G, etal. Poor stem cell harvest may not always be related to
poor mobilization: lessons gained from a mobilization study in patients with beta-thalassemia
major. Transfusion. 2017;57:1031–9.
Gopalasingam N, Thomsen AE, Folkersen L, Juhl-Olsen P, Sloth E. A successful model to
learn and implement ultrasound-guided venous catheterization in apheresis. J Clin Apher.
2017;32:437–43.
Jarisch A, Rettinger E, Sorensen J, et al. Unstimulated apheresis for chimeric antigen receptor
manufacturing in pediatric/adolescent acute lymphoblastic leukemia patients. J Clin Apher.
2020;35(5):398–405. https://doi.org/10.1002/jca.21812.
Ortiz-Maldonado V, Rives S, Castella M, etal. CART19-BE-01: a multicenter trial of ARI-0001 cell
therapy in patients with CD19(+) relapsed/refractory malignancies. Mol Ther. 2021;29:636–44.
Sörensen J, Jarisch A, Smorta C, etal. Pediatric apheresis with a novel apheresis device with elec-
tronic interface control. Transfusion. 2013;53:761–5.
Stenzinger M, Bonig H. Risks of leukapheresis and how to manage them—a non-systematic
review. Transfus Apher Sci. 2018;57:628–34.
Yakoub-Agha I, Chabannon C, Bader P, et al. Management of adults and children undergoing
CAR-T cell therapy: best practice recommendations of the EBMT and JACIE.Haematologica.
2020;105:297–316.
Key Points
• Prepare for apheresis by assessing the clinical and biological condition of
the patient.
• Discontinue treatments that can lower immune effector cell numbers and
functions.
• Tailor apheresis parameters to the patient condition.
• Tailor apheresis parameters to suit the manufacturer’s needs and
requirements.
• Tightly coordinate with the Cell Processing Facility to ensure smooth ship-
ment to the manufacturing site, in compliance with the manufacturer’s
needs and requirements.
Table 6.5 Interim storage, cryopreservation, and logistics
• Interim storage at 4–8°C
• Tisagenlecleucel: Adjust concentration to 10
8
/mL (0.5–2×10
8
); freeze as soon as feasible
but no later than 24h after collection, with DMSO as a cryoprotectant in a controlled-rate
freezer; store in LN2 (institutional SOP to be validated by the manufacturer prior to start of
operations).
• All other CAR-T cells: Ship as soon as possible at 4–8°C, following the manufacturers’
instructions and requirements.
• MAH to arrange pick-up, with temperature-controlled shipping containers of the appropriate
temperature.
• Scheduling, documentation of pick-up, hand-over, and tracking supported by MAH- supplied
specic web tools.
6 Providing the Starting Material to the Manufacturer of an Approved…

36
Open Access
This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
H. Bonig et al.

37
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_7
C. Rioufol (*)
Hospices Civils de Lyon, UCBL1, EA 3738 CICLY, Lyon, France
e-mail: [email protected]
C. Wichmann
Department of Transfusion Medicine, Cell Therapeutics and Hemostaseology, University
Hospital, LMU Munich, Munich, Germany
e-mail: Christian.W[email protected]
7
Receiving, Handling, Storage, Thawing,
Distribution, andAdministration
ofCAR-T Cells Shipped
fromtheManufacturing Facility
CatherineRioufol andChristianWichmann
Definition
In the manufacturing process for antigen receptor T cell (CAR-T cell) therapies, the
patient’s T cells acquire medicinal product status after enrichment, genetic modi-
cation, and expansion.
This pharmacologic effect results from insertion of a transgene coding for CAR,
recognizing the tumour antigen, lysing the tumour cells, and activating the immune
system see Chaps. 1, 2 and 3 in Section 1. Moreover, CAR-T cells massively expand
upon interaction with antigen- positive cells within the blood system, thereby
increasing the number of administered ATMP cells to high numbers (June et al.
2018). Due to this pharmacologic mechanism, CAR-T cells, despite their cellular
nature, are gene therapy medicinal products. Whether from patients or healthy
donors, CAR-T cells belong to the class of advanced therapy medicinal products
(ATMPs), as dened in Regulation EC N°1394/2007 of the European Parliament
and in Directive 2009/120/EC of the Council of November 13, 2007 on ATMPs,
amending Directive 2001/83/EC and Regulation N° 726/2004.
As a consequence of this medicinal status, CAR-T cells fall under the responsi-
bility of the hospital pharmacist. The manufacturer ships the released drugs to the
pharmacy of the treatment centre; the hospital pharmacist is responsible of each
step: reception, handling, storage, thawing, and dispensation (Pinturaud etal. 2018),
regardless of whether the CAR-T cells are on the market or being used experimen-
tally in a clinical trial. In hospitals with a Cell Processing Facility, the pharmacy

38
may elect to subcontract certain technical operations to the Cell Processing Facility,
as dened in an internal agreement. The overall handling and working process must
be compliant with legal requirements enforced by local and national health authori-
ties, and with the technical requirements of the drug-producing company (checked
through audits and training courses).
Workow Description of the process
Handling
– Handling of CAR-T cells according to ATMP requirements, ensuring
product safety and health care worker protection. Personal protective
equipment (PPE) to protect the handling team involved throughout the
various stages of the ‘CAR-T pathway’
– ‘CAR-T hospital pathway’ is supervised by the pharmacist; the steps are
dened by the pharmaceutical team in coordination with the medical and
nursing teams of the Haematology Department to meet objectives and ease
the patient’s care pathway
– Need of a reliable adapted quality assurance system with initial and
continuous training programs for all those involved, and periodic
assessment, in addition to the centre qualication by the pharmaceutical
laboratories
– At many centres, CAR-T cell products are managed by specialized
oncology pharmaceutical teams involved in cancer patient monitoring and
treatment, and in close contact with the oncology department
– Staff training and on-site inspection conducted by the manufacturer
– Process control through web-based communication tools, with access
provided by the manufacturer
Reception and
conformity
check
Critical steps:
– Treatment conrmation in the context of a multidisciplinary meeting that
examines that the indication is consistent with the marketing approval/
SmPC and that the patient condition is compatible with the expected safety
prole of the CAR-T cells
– The hospital pharmacist’s order is placed with the manufacturer
– Planication of the collection of the starting material through
leukapheresis at the cell collection facility (which may be operated by the
treating hospital or subcontracted to another hospital or a blood bank)
– Shipment of the starting material from the cell processing facility, which
works in close collaboration with the cell collection facility (the cell
processing facility may be operated by the treating hospital or
subcontracted to another hospital or a blood bank)
– Turnaround time of 4–6weeks between ordering and receiving to allow for
manufacturing and transportation (based on the experience of the rst
active European centres)
– Tracking of the consecutive steps of on-demand medicinal product
manufacturing on the manufacturer’s website, allowing the date and time
of reception to be known in advance to mobilize the necessary human
resources for reception without interruption of the cold chain
– Usual presentation of CAR-T cells: bag or syringe for infusion, delivered
in a dry-shipper (vapour phase nitrogen) at approximately −160°C
C. Rioufol and C. Wichmann

39
Workow Description of the process
– Reception of the dry-shipper in ventilated premises
– Conformity check at reception with reception documents (travel
documents, certicates of analysis and release, temperature logs, labels):
Cryo-shipper check: no visible damage and/or leaks
Opening of the metal cassette to fully inspect the frozen cell product
Checking the completeness and accuracy of information printed on the
CAR-T cell label: patient identity and drug identity. Proper labelling is key
to maintain the Chain of Identity/Chain of Custody throughout the
manufacturing process, up to administration of the medicinal product to
the intended recipient
– Back-up bag: whether a back-up bag is available at the manufacturer’s site
should be systematically stated in the reception documents. In case of
nonconformity detected at reception, this information is very useful for the
pharmacist and haematologists in determining the treatment strategy: a
back-up bag enables timely administration within 48h via replacement of
the defective CAR-T cells
– All retrieved information is entered on the manufacturer’s website
– Transfer of the CAR-T cells to a cryogenic recipient
– Management of out-of-range temperature or other abnormalities: storage
of CAR-T cells in quarantine and contact of the manufacturer for
instructions
– Final check of Out of Specications (OOS)
– Double control involving two members of the pharmacy team or one
member each from the pharmacy and cell processing facility for reception,
conformity checking, and transfer to the cryogenic recipient
Storage
– Storage in vapour phase nitrogen tanks. In hospitals with a cell processing
facility, storage is possible in a dedicated nitrogen tank. Respective
responsibilities are then dened in an agreement approved by the health
authority
– The cryogenic premises preferably contain several nitrogen tanks for
back-up in case of dysfunction. Having several distinct tanks also allows
CAR-T cells with market authorization to be distinguished from those
used in clinical trials or from other ATMPs. The tanks are fed by a central
lling tank, and lling should be automated and levels monitored in real
time, with a 24/7 alarm at the lower threshold. Temperature courses must
be regularly monitored, saved, and controlled
– Nitrogen storage time: approximately 6months
– Prevention of burns and hypoxia accidents in handling the CAR-T cells in
and out of the nitrogen in the cryogenic recipients: ventilated premises,
with secure access reserved to trained and retrained personnel, no access
alone to the cryogenic area, and an oculus in the door so that any incident
can be detected from the outside. Oxygen levels at the oor are recorded in
real time and displayed with visual and sound alarms at the hospital
security station. The nitrogen storage symbol is displayed. First-aid
procedures are set out, with pictograms inside and outside the premises
– Protection of the staff: use of PPE to prevent burning via nitrogen contact
(gloves with sleeves up to the elbow, protective glasses, a lab coat, and
boots). Follow institutional standard operating procedures for liquid
nitrogen handling
– Organization: stand-by duty rotation implemented for nights, weekends,
and holidays to enable intervention in case of any malfunction outside of
pharmacy opening times
Regular maintenance ensures good functioning of premises and tanks
7 Receiving, Handling, Storage, Thawing, Distribution, and Administration of CAR-T…

40
Workow Description of the process
Thawing
– After medicinal product recovery from the storage tank, a double check
(four-eyes principle) is again necessary to avoid any error in
administration: this includes a careful check of patient identity:
name, birth
date, apheresis-ID, and batch number
– Check of concordance between the product and the haematologist’s
prescription: patient identity, CAR-T denomination, administration date
– Check of the expiration date (even though CAR-T cells are stable over a
period of several months at temperatures below −160°C)
– Beginning of the thawing once the haematologist has given the green light
– Thawing operations:
Performed by the pharmaceutical team on the day of administration,
with as short a time as possible; this requires coordinated planning with
the Haematology Department
Conducted on the pharmacy premises (or in the cell processing facility
if subcontracted), after double-wrapping the bag of CAR-T cells in a
protective plastic bag in a clean room, in a dedicated 37±2°C water bath
until all ice crystals have melted in the bag. Depending on local
organization, a dry thaw method may also be used
Recommendation: double-wrap the bag in a watertight plastic bag for
thawing, to protect the bag of CAR-T cells and observe and control any
solution leakage due to accidental piercing of the original bag that may
have been overlooked at reception
After thawing, the CAR-T cells are stable at room temperature for
approximately 30 to 90min, depending on the manufacturer (please refer
to the SmPC and the manufacturer’s instructions)
– Usually, no processing step (wash, spin down, etc.) is required or allowed
– Commercial products should not be sampled
Preparation
– In case of CAR-T cells requiring processing before dispensation, injection
preparation in a pressurized preparation room with vertical laminar airow
with no air recycling is necessary to prevent the risk of microbiological
contamination of the product and to minimize risks to personnel and the
environment
Transport
to the
Haematology
departmen
t
– The interval between thawing and administration is 30 to 90min,
depending on the manufacturer, requiring precise timing, including
transport of the cells from the pharmacy to the department in a dedicated
and clearly identied container at room temperature
Warning: it is especially important to adhere to the manufacturer’s
recommended interval as it seems to be a question of the presence of
dimethyl sulfoxide (DMSO), a cryopreservation agent that impairs cell
quality and viability at room temperature, in the CAR-T cell medium (Li
etal. 2019)
– In most cases, due to the short lifespan of CAR-T cells after thawing and
the risk of patient death in case of failure to administer, transport by the
pharmacist or a member of the pharmacy staff is recommended
Dispensing in
the
Haematology
Departmen
t
– Dispensation under a ready-to-administer form from the pharmacy,
preferably to the nurse who will be in charge of injection in the
Haematology Department. Recording the dispensation time
– Check by the pharmacist that the nurse has all the specic administration
devices, notably including a non-leukodepleting in-line lter
– All material that has been in contact with the CAR-T cell product (solid and
liquid waste) should be handled and disposed of as potentially infectious
waste and genetically modied organisms (GMOs) in accordance with local
biosafety guidelines and local and national regulations
C. Rioufol and C. Wichmann

41
Administration
Five to seven days ahead of CAR-T cell injection, lymphodepleting chemotherapy
is started (Maus and June 2016). It is recommended to await reception and confor-
mity checking of the CAR-T ATMP to avoid unnecessarily reducing patient lym-
phocyte levels in cases where nonconformity prevents administration.
CAR-T cell administration is scheduled by the haematology department in coor-
dination with the pharmacy team and the Cell Processing Facility to allow for com-
pletion of the circuit under optimal conditions without delay of administration.
Patient information and consent for the entire CAR-T process, including CAR-T
cell infusion, is provided in advance by haematologists.
The cells are delivered intravenously at a 10–20mL/min infusion rate (gravity
ow) without prewarming through a peripheral or central catheter. A non-
leukodepleting in-line lter is used.
At some centres, the pharmacist is present at the bedside to respond to any
request by the nurse, such as for an extra device. Even if not physically present, the
pharmacist must remain quickly available upon request.
The hospital stay is approximately 2weeks but longer in cases of major adverse
events, such as cytokine release syndrome (CRS) or neurotoxicity, which may
require transfer to the intensive care unit. Another important responsibility that lays
with the hospital pharmacy is to ensure that two doses of tocilizumab (anti-IL-6R
antibody) are immediately available for each treated patient as per SmPC.
Patients Treated withCAR-T Cells: New Missions
fortheHospital Pharmacist
The steps along the CAR-T pathway highlight the role of the hospital pharmacist in
this therapeutic innovation.
In the future, the increasing number and variety of ATMPs may require signi-
cant changes in pharmacy organization, notably in terms of premises and equipment
(e.g., nitrogen tanks and vertical laminar airow hoods) for proper and safe han-
dling of the various types of ATMPs: somatic cell therapy medicinal products, gene
therapy medicinal products (including CAR-T cells), and other categories of
ATMPs, such as acellular gene therapy medicinal products, tissue engineering prod-
ucts, and combined medicinal products.
Key Points
• High medicinal standards of cell therapy products supervised by hospital
pharmacists.
• Cryopreservation: a strictly regulated working environment that ensures
safety of the products and requires implementation of stringent measures
to protect the physical safety of involved staff.
7 Receiving, Handling, Storage, Thawing, Distribution, and Administration of CAR-T…

42
References
Commission Directive 2009/120/EC of 14 September 2009 amending Directive 2001/83/CE of
the European Parliament and of the Council on the Community code relating to medicinal
products for human use as regards Advanced Therapy Medicinal Products (text with EEA
relevance).
June CH, O'Connor RS, Kawalekar OU, Ghassemi S, Milone MC.CAR-T cell immunotherapy
for human cancer. Science. 2018;359(6382):1361–5. https://doi.org/10.1126/science.aar6711.
Li R, Johnson R, Yu G, McKenna DH, Hubel A. Preservation of cell-based immunotherapies
for clinical trials. Cytotherapy. 2019;21(9):943–57. Epub 2019 Aug 12. PMID: 31416704;
PMCID: PMC6746578. https://doi.org/10.1016/j.jcyt.2019.07.004.
Maus MV, June CH.Making better chimeric antigen receptors for adoptive T-cell therapy. Clin
Cancer Res. 2016;22(8):1875–84. https://doi.org/10.1158/1078-0432.CCR-15-1433.
Pinturaud M, Vasseur M, Odou P.Role of the hospital pharmacist in the management of a cat-
egory of advanced therapy medicinal product: chimeric antigen receptor T-cells. Bull Cancer.
2018;105:S205–13.
Regulation (EC) N° 1394/2007 of the European Parliament and of the Council of 13 November
2007 on Advanced Therapy Medicinal Products and amending Directive 2001/83/EC and
Regulation (EC) n°726/2004 (text with EEA relevance), vol 324, 2007. http://data.europa.eu/
eli/reg/2007/1394/oj/fra
• High technical requirements, including 24/7 monitoring of the cell stor-
age site.
• Patient pathway: plan lymphodepletion chemotherapy after reception of
CAR-T cells.
• Statement in the reception document regarding whether a back-up bag is
available.
• Overall, the process is conducted within different facilities and requires
good communication skills and a multidisciplinary approach.
C. Rioufol and C. Wichmann

43
Open Access
This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
7 Receiving, Handling, Storage, Thawing, Distribution, and Administration of CAR-T…

45
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_8
J. Delgado
Department of Haematology, Hospital Clinic of Barcelona, Barcelona, Spain
e-mail: [email protected]
C. Roddie (
*)
University College London Cancer Institute, London, UK
e-mail: [email protected]
M. Schmitt
Medizinische Klinik (Krehl-Klinik), Zentrum für Innere Medizin, Klinik für Hämatologie,
Onkologie und Rheumatologie, Innere Medizin V, Heidelberg, Germany
e-mail: [email protected]
8
Point-of-Care Production ofCAR-T Cells
JulioDelgado, ClaireRoddie, andMichaelSchmitt
CAR-T cells for clinical application are classied as advanced therapy medicinal
products (ATMPs), and their manufacture is subject to laws and regulations gov-
erned by the European Medicines Agency (EMA) and by federal and regional
authorities. CAR-T cells must be manufactured to achieve good manufacturing
practice (GMP) compliance and are dened as potent products manufactured safely
according to standardized methods under closely controlled, reproducible, and
auditable conditions. BioPharma supplies the vast majority of CAR-T products for
patients, but some academic centres have developed point-of-care cGMP CAR-T
manufacturing capability, striving to uphold the same stringency of product quality
while improving patient access to CAR-T cells and streamlining the costs of ther-
apy. Point-of-care CAR-T manufacturing can only be performed in facilities with
the appropriate regulatory approvals in place.

46
GMP Vector Production
Retroviral and lentiviral vectors are the most common gene delivery methods used
in CAR-T manufacture. Viral vectors are considered an intermediate reagent by
regulatory agencies, but in manufacturing, adherence to cGMP conditions is
recommended.
Vector manufacture is conducted in grade A laminar ow cabinets in grade B
clean rooms, commonly using HEK293T packaging cell lines derived from a master
cell bank (MCB), assuming the appropriate licencing agreements with Rockfeller
University are in place. Quality control of the MCB is outlined in Table8.1 (Perpiñá
etal. 2020).
Nonviral techniques for gene transduction or gene-editing are under investiga-
tion in preclinical and early clinical trials (Prommersberger etal. 2021).
The vector manufacturing process takes 10 to 14 days and is outlined here.
Packaging cells are expanded in asks and transferred into cell culture chambers
followed by plasmid transfection using polyethylenimine. Fixed quantities of plas-
mids encoding CAR, viral envelope, and gagpol are required. Following transfec-
tion, supernatants containing the secreted vector are harvested, claried using
0.45-mm membranes, concentrated prior to dialtration and cryopreservation in
aliquots, and stored at −80°C until use. Quality measures are outlined in Table8.2
(Castellà etal. 2019).
Table 8.1 Quality control for the HEK293T master cell bank
Parameter
Method
Acceptance criteria
Appearance Visual inspection Presence of adherent cells with
thin extensions
Sterility Microbial growth Sterile
Mycoplasma PCR Absent
Adventitious viruses PCR Absent
Karyotype G-band staining Informative
Cell viability (%) after
thawing
Neubauer cell counting with
trypan blue exclusion
>70%
Table 8.2 Quality control for GMP-grade virus production
Parameter
Method
Acceptance criteria
Appearance Visual inspection Yellowish liquid solution
Viral titre Limiting dilution >3.75×10
7
TU/mL
Sterility Microbial growth Sterile
Mycoplasma PCR Absent
Identity PCR Positive
Replication-competent lentivirus Real-time PCR Absent
J. Delgado et al.

47
Manufacturing CAR-T Cells
CAR-T cell manufacturing is conducted over approximately 8–12 days in an
approved cGMP clean room facility in a closed or functionally closed system to
reduce the risk of product contamination (Roddie etal. 2019; Schubert et al. 2019;
Castellá etal. 2020).
Starting material for CAR-T cells includes CD3+ T cells derived from nonmobi-
lized leukapheresis (see Chap. 6). Mandated leukapheresis requirements of aca-
demic manufacturers for total nucleated cells (TNCs) and CD3+ T cells must be
dened; as an illustration, the Uni. Heidelberg HD-CAR-1 protocol (EudraCT No.
2016-004808-60) requires 20×10
8
TNCs and 10×10
8
CD3+ T cells, similar to
Novartis requirements for the manufacture of tisagenlecleucel. Leukapheresis mate-
rial may be cryopreserved prior to manufacture, but in a bid to shorten the manufac-
turing process, there is a trend towards using fresh leukapheresis material. CAR-T
manufacture is a stepwise process outlined in Table8.3:
Upon completion of manufacturing, CAR-T products must comply with quality
control/end-product specications stipulated in the certicate of analysis. Parameters
may vary, but CAR-T products are usually characterized for release according to
Table 8.3 CAR-T manufacturing methodology
Potential methods
Timepoint
Step 1:
T cell enrichment post-
leukapheresis (optional)
Ficoll density gradient centrifugation;
elutriation; immunomagnetic bead separation
Day 1
Step 2:
T cell activation using
synthetic antigen presenting
technologies (CD3 +/−
CD28) (required)
Soluble monoclonal antibodies; Para-magnetic
anti-CD3/CD28 antibody coated beads;
polymeric biodegradable CD3/28 incorporating
nanomatrix (TransAct™)
Days 1, 2
Step 3:
T cell stimulation (required)
IL-2, IL-7, and IL-15in the culture medium (as
per protocol) (Hoffmann etal. 2018; Gong
et al. 2019)
From day 1
onwards
Step 4:
Gene delivery/transduction
with a retroviral or lentiviral
CAR vector (required)
In some processes, retronectin or Vectofusin
®
is
used to enhance transduction (optional)
Days 2, 3
Step 5:
T cell expansion (required)
T-asks, plates or culture bags; bioreactors,
e.g., G-Rex™ ask (Wilson Wolf
Manufacturing); Xuri WAVE™ Bioreactor
(GE Life Systems); CliniMACS Prodigy™
(Miltenyi BioTec)
Days 3, 4
and onwards
Step 6:
T cell harvest and
cryopreservation (required)
The cryopreservation methodology often
mirrors processes dened for haematopoietic
cells. Methods include passive freezing
(−80°C freezer) and controlled-rate freezing
Day 8
onwards
Step 7:
CAR-T cell quality assurance
control and release testing
In-process and end of process controls are
taken to ensure the product complies with
release criteria specications
Day 8
onwards
8 Point-of-Care Production ofCAR-T Cells

48
immunophenotypic, functional, and sterility assessments (Table 8.4). An out-of-
specication (OOS) product cannot be released in the usual way, and its clinical use
is at the discretion of the treating physician in concert with the regulatory authori-
ties, informed through an OOS report.
Table 8.4 Quality control of CAR-T cell biology and microbiology
Parameter
Method
Acceptance criteria
Appearance Visual inspection Cloudy liquid solution
CAR+ cells (%)
a
Flow cytometry >20%
CD3+ cells (%) Flow cytometry >70%
Cell viability (%) Neubauer cell counting with
trypan blue exclusion
b
>70%
Sterility Microbial growth E.Ph. 2.6.1 Sterile from bacteria/fungi
Mycoplasma PCR
c
Absent
Endotoxin Chromogenic assay <0.5EU/mL
Optional/R&D
CAR/CD45RA/CCR7
For detection of TE/
TEM/TEMRA/TCM/TN
subpopulations
Flow cytometry A high proportion of immature T
cells is desirable for a long-
lasting CAR-T cell effect in the
patient
Cytotoxic potency Cr-51 release assays in tumour
CAR-T cell co-culture,
assessed by ow cytometry
>40% killing at an effector/target
ratio of 10:1 (or higher ratio) in a
4-h assay
Adventitious viruses PCR Absent
Number of transgene
copies/cell
Real-time PCR (Kunz et al.
2019; Schubert et al. 2020)
<10 (range <7–15!) copies/cell
d
a
Automated cell counters, such as Luna™, are highly recommended
b
Highly specic detection reagents (e.g., the Miltenyi Detection Reagent™) are strongly advised
to distinguish CAR-T cells from the negative fraction
c
European standards stipulate PCR methodology, in contrast to US regulations, which require
serology
d
Differs between countries and products
Summary and Key Points
• Point-of-care/decentralized CAR-T cell manufacturing has the potential to
enhance patient access to CAR-T products.
• Limitations include the requirement for local cGMP facilities/trained staff
and lack of standardization across multiple sites.
• Potential solutions include implementation of standardized, semiauto-
mated manufacturing platforms, such as the Miltenyi CliniMACS
Prodigy™, and the use of standardized release assays reported in a com-
mon format across manufacturing sites to enable the manufacture of con-
sistent, high-quality products between patients.
J. Delgado et al.

49
References
Castellà M, etal. Development of a novel anti-CD19 chimeric antigen receptor: a paradigm for
an affordable CAR-T cell production at academic institutions. Mol Ther Methods Clin Dev.
2019;12:134–44.
Castellá M, etal. Point-of-care CAR-T cell production (ARI-0001) using a closed semi-automatic
bioreactor: experience from an academic phase I clinical trial. Front Immunol. 2020;11:482.
Gong W, Hoffmann JM, Stock S, Wang L, Liu Y, Schubert ML, Neuber B, Hückelhoven-Krauss
A, Gern U, Schmitt A, Müller-Tidow C, Shiku H, Schmitt M, Sellner L.Comparison of IL-2
vs IL-7/IL-15 for the generation of NY-ESO-1-specic T cells. Cancer Immunol Immunother.
2019;68(7):1195–209. Epub 2019 Jun 8. https://doi.org/10.1007/s00262- 019- 02354- 4.
Hoffmann J-M, Schubert M-L, Wang L, Hückelhoven A, Sellner L, Stock S, Schmitt A, Kleist
C, Gern A, Loskog A, Wuchter A, Hofmann S, Ho AD, Müller-Tidow C, Dreger P, Schmitt
M.Differences in expansion potential of naive chimeric antigen receptor T cells from healthy
donors and untreated chronic lymphocytic leukemia patients. Front Immunol. 2018;8:1956.
https://doi.org/10.3389/mmu.2017.01956.
Kunz A, Gern U, Schmitt A, Neuber N, Wang L, Hückelhoven-Krauss A, Michels B, Hofmann S,
Müller-Tidow C, Dreger P, Schmitt M, Schubert M-L.Optimized assessment of qPCR-based
vector copy numbers as a safety parameter for GMP-grade CAR-T cells and monitoring of
frequency in patients. Mol Ther Methods Clin Dev. 2019;17:448–54.
Perpiñá U, etal. Cell banking of HEK293T cell line for clinical-grade lentiviral particles manufac-
turing. Transl Med Comm. 2020;5:22.
Prommersberger S, Reiser M, Beckmann J, Danhof S, Amberger M, Quade-Lyssy P, et al.
CARAMBA: a rst-in-human clinical trial with SLAMF7 CAR-T cells prepared by virus-
free Sleeping Beauty gene transfer to treat multiple myeloma. Gene Ther. 2021;28(9):560–71.
https://doi.org/10.1038/s41434-021-00254-w. Epub 2021 Apr 13. PMID: 33846552.
Roddie C, et al. Manufacturing chimeric antigen receptor T cells: issues and challenges.
Cytotherapy. 2019;21(3):327–40. https://doi.org/10.1016/j.jcyt.2018.11.009.
Schubert M-L, Schmitt A, Sellner L, Neuber B, Kunz J, Wuchter P, Kunz A, Gern U, Michels
B, Hofmann S, Hückelhoven-Krauss A, Kulozik A, Ho AD, Müller-Tidow C, Dreger P,
Schmitt M.Treatment of patients with relapsed or refractory CD19+ lymphoid disease with
T lymphocytes transduced by RV-SFG. CD19.CD28.4-1BBzeta retroviral vector: a unicen-
tre phase I/II clinical trial protocol. BMJ Open. 2019;9:e026644. https://doi.org/10.1136/
bmjopen- 2018- 026644.
Schubert ML, Kunz A, Schmitt A, Neuber B, Wang L, Hückelhoven-Krauss A, Langner S, Michels
B, Wick A, Daniel V, Müller-Tidow C, Dreger P, Schmitt M.Assessment of CAR-T cell fre-
quencies in Axicabtagene Ciloleucel and Tisagenlecleucel patients using duplex quantitative
PCR.Cancers (Basel). 2020;12(10):2820.
Open Access
This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
8 Point-of-Care Production ofCAR-T Cells

51
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_9
S. Depil (*)
Cancer Research Center of Lyon, Léon Bérard Cancer Center, University Claude Bernard
Lyon 1, Lyon, France
e-mail: [email protected].fr
W. Qasim
UCL Great Ormond Street Institute of Child Health, London, UK
e-mail: w[email protected]
9
Off-the-Shelf Allogeneic CAR-T Cells
orOther Immune Effector Cells
StephaneDepil andWaseemQasim
“Off-the-shelf” allogeneic CAR TCRαβ T cells and other immune effector cells,
such as natural killer (NK) or gamma delta (gd) T cells, can be premanufactured
from healthy donors and may offer alternatives to autologous strategies. However,
major barriers, namely HLA disparity resulting in graft versus host disease (GvHD)
and host-mediated rejection, must be addressed.
Strategies toAvoid Graft Versus Host Disease (GvHD)
Genome Edited αβTCR-Deleted T Cells
Strategies to reduce TCRαβ activity have included the use of truncated dominant-
negative CD3ζ proteins (Gilham et al. 2018), protein expression blockers (PEBLs)
(Kamiya etal. 2018), small hairpin RNA (Bunse etal. 2014), and genome editing.
Platforms for the latter have included zinc-nger nucleases (ZFN) homing endo-
nucleases/meganucleases, transcription activator-like effector nucleases (TALEN),
megaTALs, clustered regularly interspaced short palindromic repeat (CRISP/cas9),
and base editors (BEs) (Depil etal. 2020). Clinical trials of universal TCR-depleted
CAR19 T cells produced using TALENs (Servier/Allogene) have been published
(Qasim etal. 2017; Benjamin etal. 2020), and the rst applications of meganuclease

52
(Precision Bio) and CRISPR engineering were recently reported (CRISPR
Therapeutics). The manufacturing steps share common aspects of healthy donor T
cell activation with anti-CD3/CD28 antibodies, editing by electroporation of nucle-
ases and viral vector delivery of the CAR transgene. Depletion of residual TCRαβ
T cells using a magnetic bead column ensures that the carriage of potentially allore-
active T cells is kept below the threshold that might lead to GvHD.
Virus-Specific T (VST) Cells
Third-party, donor-derived VST cells have been investigated in allogeneic SCT and
appear to induce reduced levels of GvHD, presumably due to their restricted, virus-
specic, repertoire, and memory T cell phenotype. Examples include allogeneic
EBV-specic T cells transduced to express CAR19 (Curran etal. 2011) and anti-
CD30 CAR (Savoldo 2007).
Alternative Immune Effector Cells
Immune effector cells not associated with induction of GvHD, including NK cells
modied via lentiviral transduction to express CAR 19, exhibited early phase ef-
cacy in CLL (Liu etal. 2020). Similarly, iNKT or γδT cells may have advantages
against solid tumours, but clinical experience is still limited (Gentles etal. 2015).
Engineering of CAR macrophages with antitumour properties has also been
described recently (Klichinsky etal. 2020).
Strategies toAvoid Host-Mediated Rejection ofAllogeneic
Immune Cells
Beyond partial HLA matching of third-party donor cell banks, there are two main
strategies to address the risk of host-mediated rejection.
Resistance toLymphodepletion andImmunosuppression
CAR-T cells have been genome edited to become resistant to an anti-CD52
monoclonal antibody through disruption of CD52 (Benjamin etal. 2020). This
approach has the advantage of suppressing all CD52+ immune cells that can
mediate rejection, such as T, B, and NK cells, although prolonged immunosup-
pression is associated with a higher risk of serious virus reactivation. Engineering
strategies have also been used to confer resistance to calcineurin inhibitors.
Manufacturing steps can be multiplexed alongside editing of the TCR locus dur-
ing electroporation or added to the vector design and incorporated into transduc-
tion steps.
S. Depil and W. Qasim

53
Removal ofHLA forEvading Host Immunity
Removal of HLA class I molecules on the cell surface to avoid CD8
+
T cell- mediated
rejection can be achieved by disrupting the common beta 2-microglobulin gene.
This approach is currently being investigated via multiplexed editing of CAR19 T
cells (CRISPR Tx). In theory, the complete absence of HLA class I molecules may
increase NK-mediated rejection through ‘missing self’ responses, and in modelling,
this can be prevented by the expression of nonpolymorphic HLA molecules, such as
HLA-E.Rejection through recognition of HLA class II on activated T cells may be
addressed by disruption of critical transcription factors, such as CIITA and RFXANK
(Depil etal. 2020).
Manufacturing Aspects of‘Off-the-Shelf’ CARs
The majority of ‘off-the-shelf’ T cell therapies have utilized healthy donor peripheral
blood mononuclear cells (MNCs) acquired via steady-state leukapheresis, although
alternative sources, including whole blood and umbilical cord blood, may be suitable.
In the future, immune effector cells may also be derived from induced pluripotent
stem cells (iPSCs). Theoretically, a master iPSC cell line has an unlimited capability
to self-renew and can be banked and used indenitely. In general, manipulations must
be performed in a clean room setting under GMP conditions with suitable licencing
and regulatory approvals. Most cell gene transfer and genome engineering strategies
require cells to be actively in mitosis to ensure open chromatin and accessible DNA,
and activation steps are crucial early in the manufacturing process. Closed system
culture and expansion is now routine, with automation reducing labour intensive
aspects. High-quality viral vector preparations and improved electroporation steps for
genome editing and supplies of stabilized mRNA have been critical. Cryopreservation
in convenient dose formulations and efcient cold chain shipping and storage is an
essential component of premanufactured ‘off- the- shelf’ therapies.
Key Points
• Off-the-shelf allogeneic CAR-T cells devoid of signicant GvHD potential
can be manufactured.
• αβTCR-deleted CAR-T cells and CAR NK cells have successfully entered
the early clinical trial phase.
• Several strategies have been developed to avoid host-mediated rejection of
allogeneic effector cells.
9 O-the-Shelf Allogeneic CAR-T Cells orOther Immune Eector Cells

54
References
Benjamin R, etal. Genome-edited, donor-derived allogeneic anti-CD19 chimeric antigen receptor
T cells in paediatric and adult B-cell acute lymphoblastic leukaemia: results of two phase 1
studies. Lancet. 2020;396(10266):1885–94.
Bunse M, etal. RNAi-mediated TCR knockdown prevents autoimmunity in mice caused by mixed
TCR dimers following TCR gene transfer. Mol Ther. 2014;22(11):1983–91.
Curran KJ, etal. Validation of donor derived virus specic T-lymphocytes genetically modied to
target the CD19 antigen for the treatment of relapsed leukemia. Mol Ther. 2011;19:S90.
Depil S, etal. ‘Off-the-shelf’ allogeneic CAR-T cells: development and challenges. Nat Rev Drug
Discov. 2020;19(3):185–99.
Gentles AJ, etal. The prognostic landscape of genes and inltrating immune cells across human
cancers. Nat Med. 2015;21(8):938–45.
Gilham DE, et al. TCR inhibitory molecule as a promising allogeneic NKG2D CAR-T cell
approach. J Clin Oncol. 2018;36(15_suppl):e15042–2.
Kamiya T, etal. A novel method to generate T-cell receptor-decient chimeric antigen receptor T
cells. Blood Adv. 2018;2(5):517–28.
Klichinsky M, etal. Human chimeric antigen receptor macrophages for cancer immunotherapy.
Nat Biotechnol. 2020;38(8):947–53.
Liu E, etal. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl
J Med. 2020;382(6):545–53.
Qasim W, etal. Molecular remission of infant B-ALL after infusion of universal TALEN gene-
edited CAR-T cells. Sci Transl Med. 2017;9(374):eaaj2013.
Savoldo B.Blood. 2007;110(7):2620–30. https://doi.org/10.1182/blood- 2006- 11- 059139.
Open Access
This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
S. Depil and W. Qasim

Part III
Clinical Indications for CAR-T Cells

57
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_10
P. Bader (*)
Division for Stem Cell Transplantation, Immunology and Intensive Care Medicine,
Department for Children and Adolescents, University Hospital Frankfurt, Goethe University,
Frankfurt, Germany
e-mail: peter[email protected]
F. Locatelli
Department of Pediatric Hematology/Oncology and Cell and Gene Therapy, IRCCS Bambino
Gesù Children’s Hospital, Rome, Italy
e-mail: [email protected]
C. Peters
Department of Pediatrics, St. Anna Children’s Hospital, Medical University of Vienna,
Vienna, Austria
e-mail: [email protected]
10
Paediatric Acute Lymphoblastic
Leukaemia (ALL)
PeterBader, FrancoLocatelli, andChristinaPeters
Acute lymphoblastic leukaemia (ALL) is the most frequent malignant disease in
childhood and adolescence, with an annual incidence of approximately 3–4 cases
per 100,000 children under 15years of age. Multimodal chemotherapy forms the
base of current ALL treatment. Based on excellent national and international col-
laboration in consecutive prospective, randomized clinical trials, the prognosis of
childhood ALL has signicantly improved over time. Currently, up to 90% of all
paediatric patients with ALL will survive.
However, 15–20% of ALL patients eventually develop disease relapse. Of these
patients, 60–80% will achieve a second complete remission (CR) with intensive
chemotherapy regimens. Despite this high probability of obtaining a second CR,
patients with early bone marrow relapse (namely those occurring within 30months
from diagnosis) have a poor prognosis even if allogeneic stem cell transplantation
(allo-SCT) is used as consolidation therapy (Locatelli et al. 2012). According to
data from the Berlin- Frankfurt- Münster Group (BFM), patients can be grouped (S1,
S2, S3, and S4) according to the site of relapse, immune phenotype, and the time
interval between diagnosis and relapse. In S3 and S4 patients, prognosis is worse
compared to S1/S2 children, with survival rates of only 25%–30% in the whole

58
cohort of patients (von Stackelberg etal. 2011) and most recently 65% and 69% in
patients who achieved remission and could receive a transplant from a matched
donor (Peters etal. 2021).
In high-risk patients in CR 1 or in relapsed patients with low-risk proles (CR2,
S2), conventional chemotherapy followed by allo-SCT can cure up to 80% of
patients (Peters etal. 2015, 2021). In contrast, in patients who relapse after allo-
SCT, long-term survival is unlikely, and only 15% of these patients will survive the
disease (Kuhlen etal. 2018). Thus far, long-term survival is only possible through a
second allo-SCT if patients can obtain an additional CR and are t enough to receive
a second transplant. Second allo-SCT carries a considerable rate of toxicity and
mortality, and treatment lasts approximately 6–8 months; nally, approximately
30% of these patients survive (Yaniv etal. 2018).
Thus, there is an unmet medical need among children and adolescents who have
the following conditions:
– Primary refractory ALL,
– Treatment refractory relapsed ALL,
– A second relapse of their ALL, or
– Patients who relapse after allogeneic SCT and
– Patients with very high-risk ALL who should undergo allo-SCT but are not eli-
gible for the procedure for medical reasons.
Consequently, with the introduction of CD-19-directed CAR-T cell therapies,
these patients are candidates for clinical studies and nally for licencing trials of
different CAR-T cell products (Maude etal. 2018). For this patient group up to the
age of 25years, one CAR-T cell product is currently approved by the FDA and
EMA.Children, adolescents, and young adults belonging to one of the abovemen-
tioned patient groups have an indication for treatment with CAR-T cell therapies.
Whether these are the right candidates who benet most from the novel treatment
options remains to be demonstrated. Prospective studies are planned, and a few have
already begun to investigate whether the best benet of CAR-T cell treatment can
be obtained if patients were treated early in the course of the disease.
Key Points
• Patients with ALL in the second relapse, with refractory disease or who
relapse after allo-SCT may be considered for CD19 CAR-T cell therapy.
• Effective lymphodepleting chemotherapy is needed to allow expansion of
CAR-T cells.
• The level of measurable residual disease (MRD) seems to be correlated
with response and durable remission.
P. Bader et al.

59
References
Kuhlen M, Willasch AM, Dalle JH, Wachowiak J, Yaniv I, Ifversen M, Sedlacek P, Guengoer T, Lang
P, Bader P, Suiarska S, Balduzzi A, Strahm B, von Luettichau I, Hoell JI, Borkhardt A, Klingebiel
T, Schrappe M, von Stackelberg A, Glogova E, Poetschger U, Meisel R, Peters C.Outcome of
relapse after allogeneic HSCT in children with ALL enrolled in the ALL-SCT 2003/2007 trial. Br
J Haematol. 2018;180(1):82–9. Epub 2017 Nov 28. https://doi.org/10.1111/bjh.14965.
Locatelli F, Schrappe M, Bernardo ME, Rutella S.How I treat relapsed childhood acute lympho-
blastic leukemia. Blood. 2012;120(14):2807–16. Epub 2012 Aug 15. https://doi.org/10.1182/
blood- 2012- 02- 265884.
Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, Bader P, Verneris MR,
Stefanski HE, Myers GD, Qayed M, De Moerloose B, Hiramatsu H, Schlis K, Davis KL,
Martin PL, Nemecek ER, Yanik GA, Peters C, Baruchel A, Boissel N, Mechinaud F, Balduzzi
A, Krueger J, June CH, Levine BL, Wood P, Taran T, Leung M, Mueller KT, Zhang Y, Sen
K, Lebwohl D, Pulsipher MA, Grupp SA. Tisagenlecleucel in children and young adults
with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439–48. PMID: 29385370;
PMCID: PMC5996391. https://doi.org/10.1056/NEJMoa1709866.
Peters C, Schrappe M, von Stackelberg A, Schrauder A, Bader P, Ebell W, Lang P, Sykora KW,
Schrum J, Kremens B, Ehlert K, Albert MH, Meisel R, Matthes-Martin S, Gungor T, Holter
W, Strahm B, Gruhn B, Schulz A, Woessmann W, Poetschger U, Zimmermann M, Klingebiel
T. Stem-cell transplantation in children with acute lymphoblastic leukemia: a prospective
international multicenter trial comparing sibling donors with matched unrelated donors-the
ALL-SCT-BFM-2003 trial. J Clin Oncol. 2015;33(11):1265–74. Epub 2015 Mar 9. https://doi.
org/10.1200/JCO.2014.58.9747.
Peters C, Dalle JH, Locatelli F, Poetschger U, Sedlacek P, Buechner J, Shaw PJ, Staciuk R, Ifversen
M, Pichler H, Vettenranta K, Svec P, Aleinikova O, Stein J, Güngör T, Toporski J, Truong TH,
Diaz-de-Heredia C, Bierings M, Arifn H, Essa M, Burkhardt B, Schultz K, Meisel R, Lankester
A, Ansari M, Schrappe M, IBFM Study Group, von Stackelberg A, IntReALL Study Group,
Balduzzi A, I-BFM SCT Study Group, Corbacioglu S, EBMT Paediatric Diseases Working
Party, Bader P.Total body irradiation or chemotherapy conditioning in childhood ALL: a mul-
tinational, randomized, noninferiority phase III study. J Clin Oncol. 2021;39(4):295–307. Epub
2020 Dec 17. https://doi.org/10.1200/JCO.20.02529.
von Stackelberg A, Völzke E, Kühl JS, Seeger K, Schrauder A, Escherich G, Henze G, Tallen G,
ALL-REZ BFM Study Group. Outcome of children and adolescents with relapsed acute lym-
phoblastic leukaemia and non-response to salvage protocol therapy: a retrospective analysis of
the ALL-REZ BFM Study Group. Eur J Cancer. 2011;47(1):90–7. Epub 2010 Oct 20. https://
doi.org/10.1016/j.ejca.2010.09.020.
Yaniv I, Krauss AC, Beohou E, Dalissier A, Corbacioglu S, Zecca M, Afanasyev BV, Berger M,
Diaz MA, Kalwak K, Sedlacek P, Varotto S, Peters C, Bader P.Second hematopoietic stem cell
transplantation for post-transplantation relapsed acute leukemia in children: a retrospective
EBMT-PDWP study. Biol Blood Marrow Transplant. 2018;24(8):1629–42. Epub 2018 Mar 13.
Open Access
This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
10 Paediatric Acute Lymphoblastic Leukaemia (ALL)

61
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_11
E. Jacoby · A. Nagler (*)
Sheba Medical Center, Tel Aviv University, Ramat Gan, Israel
e-mail: [email protected].il; [email protected].il
N. Gökbuget
Department of Haematology/Oncology, Goethe University,
Frankfurt am Main, Hessen, Germany
e-mail: [email protected]
11
Adult Acute Lymphoblastic Leukaemia
EladJacoby, NicolaGökbuget, andArnonNagler
ALL is a malignancy of lymphoid progenitor cells, with a bimodal incidence, peak-
ing in early childhood and in older age. In children, ALL tends to have an excellent
prognosis, with more than 85% of patients achieving long-term survival. The out-
come of younger adults has improved considerably as well. However, overall sur-
vival decreases with age (Dores etal. 2012), partially due to the different genetic
background of adult ALL, with a higher proportion of Philadelphia chromosome-
positive (Ph+) ALL and Ph-like and KMT2A rearrangements in comparison to
childhood ALL (Iacobucci and Mullighan 2017). The introduction of paediatric-
inspired regimens has improved outcomes in adults, but these regimens are less
tolerated in older patients (Curran and Stock 2015).
The standard upfront therapy for ALL includes corticosteroids, multiagent che-
motherapy, antimetabolite therapy, and intrathecal therapy. Following induction,
consolidation and maintenance therapy are initiated. In high-risk cases, allogeneic
haematopoietic stem-cell transplantation (allo-HSCT) is considered during the rst
remission. Adults with relapsed ALL have a poor chance of achieving remission
with chemotherapy (Frey and Luger 2015). Novel agents, such as inotuzumab ozo-
gamicin, an antibody–drug conjugate targeting CD22, and blinatumomab, a bispe-
cic engager targeting CD19 and CD3, improve remission rates, but overall survival
remains poor (Kantarjian etal. 2016, 2017). Relapse therapy is usually followed by
allo-HSCT if not performed earlier. ALL cases refractory to two or more lines of
therapy can be considered for CAR-T cell therapy.

62
CAR-T Cell Therapy forAdult ALL
Currently, in 2021, no regulatory agency has approved a CAR-T cell product for
adult ALL patients above 25years of age. Young adults aged 18–25 were included
in the pivotal ELIANA study and are eligible for tisagenlecleucel (Maude et al.
2018). Other single-institutional studies also included young adults in a paediatric-
focused study. Only a few groups have reported clinical trials in adults with ALL
(Table11.1). Most trials include small patient numbers, usually younger adults, and
may represent selected patient populations. Remission rates across trials are high,
with more than 70% of patients achieving complete remission, regardless of cytoge-
netic background, prior therapies and age. Occasionally, response rates are reported
as intent-to-treat, referring to all included patients in contrast to only those receiving
CAR-T cell therapy.
Toxicity has been a signicant issue in all trials, and fractionation of the dose by
administration of a partial dose on Day 0 and the remainder after several days was
shown to be safer (Frey etal. 2019; Park etal. 2018). Several groups also adminis-
tered lower doses to patients with a high disease burden to prevent toxicity (Roddie
etal. 2020). Alternative approaches to enhance safety include earlier administration
of tocilizumab and low-dose steroids (Gardner etal. 2019; Kadauke etal. 2021; Liu
etal. 2020). Using a novel low afnity CD19 CAR-T cell was also associated with
lower toxicity (Ghorashian etal. 2019; Roddie etal. 2020).
The prognostic factors that are associated with higher remission rates and better
outcome in adult ALL include lower disease burden, as assessed by the blast count
in bone marrow; lower LDH; and higher platelet count prior to lymphodepletion
(Hay etal. 2019; Park etal. 2018). Due to the time delay between the detection of
relapse and infusion of CAR-T cells, in many cases, it is necessary to deliver bridg-
ing therapy. The optimal regimens need to be dened.
Assessing the leukaemia burden before CAR-T-infusion and after potential
bridging therapies is recommended because the outcome of patients with a high
disease burden is inferior to that of those without persistent disease or minimal
residual disease (MRD) only. The results may be inferior in ALL patients previ-
ously treated with blinatumomab (Pillai etal. 2019), although this may represent
a selection of more resistant patients. TP53 mutations were associated with a
worse outcome. Additionally, conditioning with udarabine and cyclophospha-
mide was superior to cyclophosphamide alone in adults, similar to ndings in
children.
Many trials report MRD status determined by ow cytometry post CAR-T
cell therapy, showing that almost all remissions are (based on ow-cytometry)
MRD negative. Molecular detection of MRD via PCR or next-generation
sequencing (NGS) is more sensitive, and NGS-MRD negativity after CAR-T
cells has been shown to be associated with an improved long-term outcome
(Hay etal. 2019).
E. Jacoby et al.

63
Table 11.1 Clinical trials reporting the outcome of adult ALL treated with CAR-T cells
Group
n LD Construct Dose CR (%)
MRD neg
of CR
Consolidative
therapy
Comments
U.Penn
(Frey
etal.)
35 Cy (300×6,
n=25), Flu/Cy
(n=5), other
(n=3), none
(n=2)
FMC63-41BBz 5×10
8
single/
fractionated
24 (69) 100%
(ow)
9 HSCT, 15
none
2 y OS 47%;
fractionation of dose is
safer
FHCRC
(Hay etal.)
53 Cyclophosphamide
(n=11) vs. u/cy
(n=42)
FMC63-41BBz
at CD4:CD8
prespecied
ratios
2×10
5
/kg
(n=33),
2×10
6
/kg
(n=20)
45 (85) 100%
(ow),
71%
(20/28
NGS)
18 HSCT, 27
none
Low LDH and high
platelet levels pre-LD
improve outcome;
median OS 20months
in responders
MSKCC
(Park
etal.)
53 Cyclophosphamide
(3g/m
2
, n=43),
Flu/Cy (n=10)
MSKCC-28z 1–3×10
6
/kg 44 (83%) 72% (32
of 44)
17 HSCT, 26
none, 1 other
Median survival
12.9months
Beijing
(Dai etal.)
6 Flu/Cy CD19/22 (m971/
FMC63)-41BBz
1.7–3×10
6
/kg 6 (100) 100% 3 relapsed, short
follow-up time
UCL
AUTO1
(Roddie
etal.)
19 Flu/Cy CAT-41BBz 10–100×10
6
16 (84) 100% 2 HSCT, 14
none
Lu Daopei
(Zhang
etal.)
110 (39
adults)
Flu/Cy CD19-28z
(n=21),
CD19-41BBz
(n=89)
1–10×10
6
/kg 102 (92) 94% 75 HSCT, 27
none
Worse outcome with
TP53 mutations
11 Adult Acute Lymphoblastic Leukaemia

64
Consolidation After CAR-T Cell Therapy
Despite durable CAR-T cells being applied as denitive therapy for relapsed ALL
in children, adult data are controversial. Outcomes were not improved by allo-
HSCT in patients treated with CD28-based CAR-T cells, which have short-term
persistence (Park etal. 2018). In contrast, adults treated with CLT019 (Frey etal.
2019) had better outcomes if transplanted during CR after CAR-T cell therapy.
Several centres recommend allo-HSCT for adult ALL patients following CAR-T
cell therapy even in the presence of MRD-negative remission (Hay et al. 2019;
Zhang etal. 2020; Zhao etal. 2020). Patients with molecular MRD positivity fol-
lowing CAR-T cell therapy, patients with rapid loss of CAR-T cells, and patients
who have not received a previous HSCT are candidates for consolidative HSCT
(Jacoby 2019; Jiang etal. 2020).
Relapse After CAR-T Cell Therapy
Relapse after CAR-T cell therapy occurs in 30–50% of patients. In instances of
durable CAR-T cells, there is a higher probability that relapsed ALL will not express
CD19, occurring in up to 40% of cases. If CAR-T cells are lost early, CD19 expres-
sion may be preserved. A second dose of CAR-T cells led to rare responses in
patients with ALL who relapsed after CAR-T cell therapy or were refractory to this
treatment (Gauthier etal. 2020). Other therapies, such as novel antibody-based or
CAR-T cells targeting other antigens, are optional.
References
Curran E, Stock W. How I treat acute lymphoblastic leukemia in older adolescents and young
adults. Blood. 2015;125:3702–10. https://doi.org/10.1182/blood- 2014- 11- 551481.
Dores GM, Devesa SS, Curtis RE, Linet MS, Morton LM.Acute leukemia incidence and patient
survival among children and adults in the United States, 2001–2007. Blood. 2012;119:34–43.
https://doi.org/10.1182/blood- 2011- 04- 347872.
Key Points
• No CAR-T cell product is approved for patients with ALL older than
25years.
• Clinical trial data for adult ALL are limited.
• CAR-T cells appear to me more effective and tolerated better if used in the
MRD setting of ALL.
• The use of consolidative HSCT after CAR-T cells in adults is still a matter
of debate.
E. Jacoby et al.

65
Frey NV, Luger SM. How I treat adults with relapsed or refractory Philadelphia chromosome-
negative acute lymphoblastic leukemia. Blood. 2015;126:589–96. https://doi.org/10.1182/
blood- 2014- 09- 551937.
Frey NV, Shaw PA, Hexner EO, Pequignot E, Gill S, Luger SM, et al. Optimizing chimeric
antigen receptor T-cell therapy for adults with acute lymphoblastic leukemia. J Clin Oncol.
2019;38:415–22.
Gardner RA, Ceppi F, Rivers J, Annesley C, Summers C, Taraseviciute A, etal. Preemptive miti-
gation of CD19 CAR-T cell cytokine release syndrome without attenuation of antileukemic
efcacy. Blood. 2019;134:2149–58. https://doi.org/10.1182/blood.2019001463.
Gauthier J, Bezerra ED, Hirayama AV, Fiorenza S, Sheih A, Chou CK, etal. Factors associated
with outcomes after a second CD19-targeted CAR-T cell infusion for refractory B cell malig-
nancies. Blood. 2020;137:323–35. https://doi.org/10.1182/blood.2020006770.
Ghorashian S, Kramer AM, Onuoha S, Wright G, Bartram J, Richardson R, et al. Enhanced
CAR-T cell expansion and prolonged persistence in pediatric patients with ALL treated
with a low- afnity CD19 CAR. Nat Med. 2019;25(9):1408–14. https://doi.org/10.1038/
s41591- 019- 0549- 5.
Hay KA, Gauthier J, Hirayama AV, Voutsinas JM, Wu Q, Li D, et al. Factors associated with
durable EFS in adult B-cell ALL patients achieving MRD-negative CR after CD19 CAR-T
cells. Blood. 2019;133:1652–63. https://doi.org/10.1182/blood- 2018- 11- 883710.
Iacobucci I, Mullighan CG. Genetic basis of acute lymphoblastic leukemia. J Clin Oncol.
2017;35:975–83. https://doi.org/10.1200/JCO.2016.70.7836.
Jacoby E.The role of allogeneic HSCT after CAR-T cells for acute lymphoblastic leukemia. Bone
Marrow Transplant. 2019;54:810–4. https://doi.org/10.1038/s41409- 019- 0604- 3.
Jiang H, Hu Y, Mei H.Consolidative allogeneic hematopoietic stem cell transplantation after chi-
meric antigen receptor T-cell therapy for relapsed/refractory B-cell acute lymphoblastic leuke-
mia: who? when? why? Biomark Res. 2020;8:66. https://doi.org/10.1186/s40364- 020- 00247- 8.
Kadauke S, Myers RM, Li Y, Aplenc R, Baniewicz D, Barrett DM, etal. Risk-adapted preemptive
tocilizumab to prevent severe cytokine release syndrome after CTL019 for pediatric B-cell
acute lymphoblastic leukemia: a prospective clinical trial. J Clin Oncol. 2021;39(8):920–30.
https://doi.org/10.1200/jco.20.02477.
Kantarjian HM, DeAngelo DJ, Stelljes M, Martinelli G, Liedtke M, Stock W, etal. Inotuzumab
Ozogamicin versus standard therapy for acute lymphoblastic leukemia. N Engl J Med.
2016;375:740–53. https://doi.org/10.1056/NEJMoa1509277.
Kantarjian H, Stein A, Gökbuget N, Fielding AK, Schuh AC, Ribera J-M, etal. Blinatumomab ver-
sus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med. 2017;376:836–47.
https://doi.org/10.1056/NEJMoa1609783.
Liu S, Deng B, Yin Z, Pan J, Lin Y, Ling Z, et al. Corticosteroids do not inuence the ef-
cacy and kinetics of CAR-T cells for B-cell acute lymphoblastic leukemia. Blood Cancer
J. 2020;10:20–3. https://doi.org/10.1038/s41408- 020- 0280- y.
Maude SL, Latesch T, Buechner J, Rives S, Boyer M, Bittencourt H, etal. Tisagenlecleucel in chil-
dren and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378:439–48.
https://doi.org/10.1056/NEJMoa1709866.
Park JH, Rivière I, Gonen M, Wang X, Sénéchal B, Curran KJ, etal. Long-term follow-up of CD19
CAR-therapy in acute lymphoblastic leukemia. N Engl J Med. 2018;378:449–59. https://doi.
org/10.1056/NEJMoa1709919.
Pillai V, Muralidharan K, Meng W, Bagashev A, Oldridge DA, Rosenthal J, etal. CAR-T cell ther-
apy is effective for CD19-dim B-lymphoblastic leukemia but is impacted by prior blinatumomab
therapy. Blood Adv. 2019;3:3539–49. https://doi.org/10.1182/bloodadvances.2019000692.
Roddie C, O’Reilly MA, Marzolini MAV, Wood L, Dias J, Cadinanos Garai A, etal. ALLCAR19:
updated data using AUTO1, a novel fast-off rate CD19 CAR in relapsed/refractory B-cell acute
lymphoblastic Leukaemia and other B-cell malignancies. Blood. 2020;136:3–4. https://doi.
org/10.1182/blood- 2020- 137768.
11 Adult Acute Lymphoblastic Leukaemia

66
Zhang X, Lu XA, Yang J, Zhang G, Li J, Song L, etal. Efcacy and safety of anti-CD19 CAR-T
cell therapy in 110 patients with B-cell acute lymphoblastic leukemia with high-risk features.
Blood Adv. 2020;4:2325–38. https://doi.org/10.1182/bloodadvances.2020001466.
Zhao H, Wei J, Wei G, Luo Y, Shi J, Cui Q, etal. Pre-transplant MRD negativity predicts favorable
outcomes of CAR-T therapy followed by haploidentical HSCT for relapsed/refractory acute
lymphoblastic leukemia: a multi-center retrospective study. J Hematol Oncol. 2020;13:1–13.
https://doi.org/10.1186/s13045- 020- 00873- 7.
Open Access
This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
E. Jacoby et al.

67
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_12
B. Glass
Clinic for Haematology, Oncology, Tumour Immunology, and Palliative Care, Helios
Klinikum Berlin-Buch, Berlin, Germany
e-mail: [email protected]
M. J. Kersten (
*)
Department of Hematology, Amsterdam University Medical Centers, University of
Amsterdam, Amsterdam, The Netherlands
e-mail: [email protected]
12
Diffuse Large B Cell Lymphoma
andPrimary Mediastinal Lymphoma
BertramGlass andMarieJoséKersten
The outcome of patients with large B cell lymphoma (LBCL) who did not respond
to a classical immunochemotherapy regimen at any time or relapsed within 1 year
following chemoimmunotherapy is poor. The Scholar-One-Study showed long-term
event-free survival for less than 20% of these patients (Crump etal. 2017). The
introduction of chimeric antigen receptor T cell therapy (CAR-T) is a substantial
advancement for these patients, offering long-term remission and a curative pros-
pect for 30 to 40% of patients (summarized in Table12.1), (Abramson etal. 2020;
Neelapu etal. 2017; Schuster etal. 2019b). To date, in Europe, two products (axi-
cabtagene ciloleucel and tisagenlecleucel) have been licenced by the European
Medical Agency, and a third product (lisocabtagene maraleucel) will become avail-
able in 2022. All these products are licenced for patients who have failed at least two
prior lines of systemic therapy. This initially denes, however broad, a range of
possible situations in which the application of CART is indicated. The following
considerations may help to further dene the patient population that should be
offered CAR-T cells as the next line of treatment. Recently the results of three ran-
domized phase III clinical studies comparing CD19-CART with standard of Care in
transplant eligible patients were reported. The BELINDA-Trial using Tisa-cel did
not reach its primary endpoint EFS (Bishop et al. 2021). Two of the studies,
ZUMA-7 using the construct Axi-cel and TRANSFORM using Liso-cel were posi-
tive for their primary endpoint EFS as well as for the key secondary endpoints PFS
and ORR (Kamdar et al. 2021; Locke et al. 2021). In both studies a strong numerical

68
trend towards a positive result regarding OS was observed with Hazard ratios of
0.72 and 0.51, respectively. TRANSFORM had a short median observation time so
the results regarding OS were immature. In both studies showed signicant advan-
tages in quality of life for CART therapy over ASCT. Anti-CD19 CART therapy
with one of these compounds should be considered standard of care in transplant-
eligible patients as second line therapy in R/R LBCL.
Patient Population toConsider: Lymphoma-Specific Aspects
Treatment History
Depending on clinical risk factors, between 5 and 50% of LBCL patients may fail
standard rst-line immunochemotherapy (Coifer etal. 2010; Cunningham etal.
2013; Pfreundschuh etal. 2011; Schmitz etal. 2012). Overall, 30–40% of patients
will need salvage treatment. Patients eligible for high-dose chemotherapy currently
receive a platinum-containing salvage protocol, followed by high- dose chemother-
apy and autologous stem cell transplantation (autoSCT). In short, the remission rate
varies between 35 and 50%, and with another approximately 50% failure rate after
autoSCT, the overall long-term event-free survival rate is in the range of 25%. Thus,
approximately 75% of younger patients with relapsed/refractory aggressive B cell
lymphoma are theoretically eligible for CAR-T cell therapy. All young, t patients
without remission after salvage therapy in second- line situations should be consid-
ered for CAR-T cell therapy.
Elderly patients with relapsed or refractory LBCL have poor results with
second- line therapy. An analysis of the secondary overall survival of patients in a
large randomized rst-line study revealed that patients with primary refractory
disease never achieved long-term remission, and the median overall survival was
less than 6 months (Glass etal. 2017). In 20–30% of patients, induction of remis-
sion is possible with platinum- or bendamustine-containing regimens, such as
Table 12.1 Results of pivotal trials of anti-CD19 CAR-T cell therapy
References
Axi-cel
Zuma1
Tisa-cel
Juliet
Liso-cel
Transcend
n (pts infused vs. apheresed) 101/111 (91%) 115/167 (69%) 294/344 (85%)
a
Age 58 (23–76) 56 (22–76) 63 (18–86)
Prior lines of therapy (median) 3 (1–8) 2 (1–6) 3 (1–8)
Patients refractory (%) 78 (77%) 61 (55%) 181 (67%)
Bridging therapy (%) 0 92 59
ORR (%) 82 52 73
CR (%) 54 32 53
PFS (%) 41 31 44 (1 year)
TRM (%) 4 0 3
a
25 patients received a non-conforming product
B. Glass and M. J. Kersten

69
R- gemcitabine-oxaliplatin; however, due to the lack of effective consolidative
therapy, the long-term outcome was poor in all series (Dhanapal et al. 2017;
Franch-Sarto etal. 2019). In the three pivotal CD19 CAR-T trials, 23–42% of
patients were ≥65years old, and there are currently no indications that the ef-
cacy of CAR-T cells in terms of remission rate and progression-free survival is
inferior for elderly patients (Neelapu, Schuster, Abramson). In a real-world series,
the percentage of elderly patients was higher, and there was some evidence of
increased toxicity, especially neurotoxicity, but the efcacy appeared to be at last
equal (Pasquini CIBMTR, Nastoupil).
Therefore, the option of CAR-T cell therapy must be discussed for most trans-
plant ineligible but t patients with relapsed or refractory aggressive B cell lym-
phoma after 2 prior lines of systemic therapy.
Remission Status and Tumour Bulk Prior to CAR-T Cell Infusion
CAR-T cells have been evaluated in patients with refractory and early relapsed dis-
ease. In pivotal studies of the three licenced CAR-T products, the fraction of refrac-
tory patients varied between 52 and 79%. Being refractory to the last line of
chemotherapy was not a signicant prognostic factor in these studies. Therefore, in
contrast to autologous or allogeneic SCT, being in remission is not a prerequisite for
the application of CAR-T therapy. However, pivotal clinical studies and the rst
real-world evidence reports identify high tumour volume, reected by the sum of
product diameters (SPD) or simply an elevated LDH, prior to lymphodepleting
therapy as a signicant negative prognostic factor for the ongoing complete response
rate or PFS (Nastoupil etal. 2020), with a hazard ratio of 3.0. In addition, patients
with rapidly progressing disease often do not respond to attempts at bridging ther-
apy, and the need for systemic bridging therapy is a negative predictor for response
and survival.
When discussing the value of CAR-T cell therapy for an individual patient, this
parameter—disease control prior to CAR-T cell infusion—must be considered.
However, this parameter also has an impact on the results of any alternative treat-
ment, and at which point CAR-T cell therapy cannot achieve long-term remission
and should not be offered is still a matter of debate.
Histology
The majority of patients in ZUMA-1, JULIET, and TRANSCEND had DLBCL
(76%, 80% and 64%); transformed FL was present in 16–22% of cases, and only
a small number of patients with PMBCL (8% in ZUMA-1 and 6% in TRANSCEND)
were included. Data on DH/TH lymphoma were not available for all patients.
There were no signicant differences in response rates or PFS in specic
subgroups.
12 Diuse Large B Cell Lymphoma andPrimary Mediastinal Lymphoma

70
Patient Population toConsider: Patient-Specific Aspects
CAR-T cell therapy can lead to unusual and sometimes severe acute toxicities, with
cytokine release syndrome (CRS) and immune effector cell-associated neurotoxic-
ity (ICANS) being the most important. These toxicities typically occur between
Day 2 and Day 10 after infusion and may persist for several days to weeks.
Treatment-related mortality is a rare event, and in most cases, the toxicity is revers-
ible. Other important side effects include severe neutropenia, which may last for
several weeks or months, long-term B cell depletion, and hypogammaglobulinae-
mia, which is an on-target off-tumour toxicity. Compared to the morbidity and mor-
tality associated with other treatment modalities applied in this situation, such as
allogeneic SCT, the impact of CAR-T cell-associated toxicity on the overall out-
come is moderate. It has been claimed that the good results of CAR-T therapy in
terms of toxicity are due to the strict eligibility criteria of the pivotal studies, but
recent analyses of data from real-world application of CAR-T therapy showed that
efcacy and toxicity were similar, even if elderly and more comorbid patients were
treated (Nastoupil etal. 2020). The risk factors for TRM after CAR-T cell therapy
are not well dened, and conclusions from other treatment options might be difcult
to transfer to CAR-T cell therapy. The best approximation might be the use of the
high-dose chemotherapy comorbidity index and its results for patients undergoing
autologous SCT.In such an analysis, the HDT-correlated NRM ranged from 3.3 to
7.7% (Berro etal. 2017) after 1year, which is in line with what has been observed
after CAR-T cell therapy (therapy-related mortality <5%).
Alternative Treatments
In the past, allogeneic stem cell transplantation was the only option for consolida-
tion in chemorefractory patients after additional salvage therapy. Recently, anti-
body–drug conjugates and bispecic antibodies have shown interesting results in
the patient population discussed here. Polatuzumab vedotin, the rst drug in these
groups, has been licenced in Europe and the USA.Many other drugs may enter the
arena in the near future. Integration of CAR-T cell therapy with these new treatment
options will be a major task in the future.
• Allogeneic stem cell transplantation
In the past, alloSCT has mostly been used in patients who relapse after autolo-
gous stem cell transplantation, and data have been reported from international
registries (Fenske etal. 2016; van Kampen etal. 2011). One prospective clinical
trial in patients with high-risk aggressive lymphoma showed a PFS and OS of
39% and 40%, respectively, after 4years (Glass etal. 2014). Taking the potential
bias of indirect comparison of treatment results between studies and registry data
into account, it can be grossly stated that in many series of allogeneic stem cell
transplantation, the overall results in terms of progression-free survival may be
comparable to that of CAR-T therapy; however, the balance between the anti-
B. Glass and M. J. Kersten
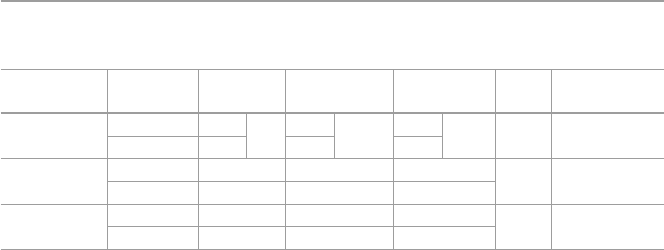
71
lymphoma effect and toxicity is dramatically different from that of CAR-T ther-
apy. Treatment-related mortality and morbidity are dramatically higher after
allogeneic stem cell transplantation in any age group of patients. Thus, currently,
even in patients eligible for alloSCT, CAR-T cells should be offered rst.
Allogeneic SCT remains a treatment modality that should be considered in
patients failing CART therapy, provided they respond to salvage treatment.
• Antibody drug conjugates
Polatuzumab vedotin is the rst ADC that has been licenced for transplant-
ineligible patients with DLBCL after failure of at least one prior therapy. In the
pivotal randomized phase II study (Sehn etal. 2020a) and the recently reported
expansion cohort (Sehn etal. 2020b), the patient population treated was compa-
rable to the populations in most of the CAR-T cell studies. The best overall
response rate was 57.9%, with a CR rate of 52.6%. Some of the responses seem
to be ongoing after the end of treatment. With the limited number of patients and
the limited observation time, it can be estimated that approximately 15–20% of
patients might be in ongoing remission after 2years. Thus, the potential to serve
as a curative treatment is small if it exists at all, and a longer follow-up time and
validation in other prospective clinical trials are warranted. ADCs, such as
polatuzumab vedotin, might be good candidates for achieving control of the dis-
ease prior to CAR-T cell application in a so-called bridging approach.
Polatuzumab is licenced in combination with bendamustine, a drug with excep-
tionally high T cell toxicity. The application of bendamustine should be avoided
prior to apheresis of autologous T cells for CAR-T cell production.
• Bispecic antibodies
A number of bispecic, T cell-engaging antibodies using the CD20 antigen as
a lymphoma-specic target and CD3 as a T cell binding site have been reported
with very encouraging results (Table12.2) (Bannerji etal. 2020; Hutchings etal.
2020a,b; Schuster et al. 2019a). The response rates are high for some of the
agents, and the toxicity is quite limited. However, the observation time is still
short, and data on PFS are not yet available. There is an indication that the DOR
of patients achieving CR is particularly good thus far and in the same range as
that observed in trials of CAR-T cells. Bispecic antibodies may have the poten-
Table 12.2 Results of prospective randomized trials of second-line treatment for aggres-
sive B-NHL
Study
Salvage
regimen ORR (%) PFS/EFS OS FU
References
CORAL R-DHAP 62.8 51
a
42% 21%
a
EFS
51% 40%
a
3
years
Gisselbrecht
etal. (2010)
R-ICE 63.5 31% 47%
NCIC-CTG
LY.12
b
R-DHAP 44.1 26% EFS 39% 4
years
Crump etal.
(2014)
R-GDP 45.1 26% EFS 39%
ORCHARD R-DHAP 42 26% 38% 2
years
van Imhoff
etal. (2017)
O-DHAP 38 24% 41%
a
Results in patients receiving rituximab as rst-line therapy
b
The study included 8% of patients with T cell lymphoma; 67% of patients received prior rituximab
12 Diuse Large B Cell Lymphoma andPrimary Mediastinal Lymphoma

72
tial to induce long-lasting remissions without further consolidation, but this
treatment is often given until progression or toxicity occurs. Once available in
routine practice, their differential indication for treatment compared to CAR-T
cells will become an important clinical challenge and should preferably be inves-
tigated in head-to-head clinical trials.
References
Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang M, Arnason J, etal. Lisocabtagene
maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND
NHL 001): a multicentre seamless design study. Lancet. 2020;396:839–52.
Bannerji R, Allan JN, Arnason JE, Brown JR, Advani R, Ansell SM, et al. Odronextamab
(REGN1979), a human CD20 x CD3 bispecic antibody, induces durable, complete responses
in patients with highly refractory B-cell non-Hodgkin lymphoma, including patients refractory
to CAR-T therapy. Blood. 2020;136:42–3.
Berro M, Arbelbide JA, Rivas MM, Basquiera AL, Ferini G, Vitriu A, etal. Hematopoietic cell
transplantation-specic comorbidity index predicts morbidity and mortality in autologous stem
cell transplantation. Biol Blood Marrow Transplant. 2017;23:1646–50.
Bishop MR, Dickinson M, Purtill D, Barba P, Santoro A, Hamad N, et al. Second-Line tisagenle-
cleucel or standard care in aggressive B-Cell lymphoma. N Engl J Med. 2021. Dec 14, Online
ahead of print.
Coifer B, Thieblemont C, Van Den Neste E, Lepeu G, Plantier I, Castaigne S, etal. Long-term
outcome of patients in the LNH-98.5 trial, the rst randomized study comparing rituximab-
CHOP to standard CHOP chemotherapy in DLBCL patients: a study by the Groupe d’Etudes
des Lymphomes de l’Adulte. Blood. 2010;116:2040–5.
Crump M, Kuruvilla J, Couban S, Macdonald DA, Kukreti V, Kouroukis CT, etal. Randomized
comparison of gemcitabine, dexamethasone, and cisplatin versus dexamethasone, cytarabine,
and cisplatin chemotherapy before autologous stem-cell transplantation for relapsed and refrac-
tory aggressive lymphomas: NCIC-CTG LY.12. J Clin Oncol. 2014;32:3490–6.
Crump M, Neelapu SS, Farooq U, Van Den Neste E, Kuruvilla J, Westin J, etal. Outcomes in
refractory diffuse large B-cell lymphoma: results from the international SCHOLAR-1 study.
Blood. 2017;130:1800–8.
Cunningham D, Hawkes EA, Jack A, Qian W, Smith P, Mouncey P, etal. Rituximab plus cyclo-
phosphamide, doxorubicin, vincristine, and prednisolone in patients with newly diagnosed dif-
fuse large B-cell non-Hodgkin lymphoma: a phase 3 comparison of dose intensication with
14-day versus 21-day cycles. Lancet. 2013;381:1817–26.
Key Points
• Patients with LBCL, including transformed FL and PMBCL, should be
considered for CD19 CAR-T cell therapy in cases of relapsed/refractory
disease after ≥2 lines of systemic therapy.
• There is no upper age limit, but patients should full eligibility criteria in
terms of tness, cardiac function, and other organ functions.
• Patients with high LDH and rapidly progressing disease are less likely to
benet.
B. Glass and M. J. Kersten

73
Dhanapal V, Gunasekara M, Lianwea C, Marcus R, De Lord C, Bowcock S, etal. Outcome for
patients with relapsed/refractory aggressive lymphoma treated with gemcitabine and oxaliplatin
with or without rituximab; a retrospective, multicentre study. Leuk Lymphoma. 2017;58:1–9.
Fenske TS, Ahn KW, Graff TM, DiGilio A, Bashir Q, Kamble RT, etal. Allogeneic transplantation
provides durable remission in a subset of DLBCL patients relapsing after autologous transplan-
tation. Br J Haematol. 2016;174:235–48.
Franch-Sarto M, Sorigue M, Lopez L, Moreno M, Ribera JM, Sancho JM. Overall survival in
patients with relapsed/refractory high grade B-cell lymphomas treated with gemcitabine, oxali-
platin with or without rituximab. Leuk Lymphoma. 2019;60:3324–6.
Gisselbrecht C, Glass B, Mounier N, Singh GD, Linch DC, Trneny M, etal. Salvage regimens
with autologous transplantation for relapsed large B-cell lymphoma in the rituximab era. J Clin
Oncol. 2010;28:4184–90.
Glass B, Hasenkamp J, Wulf G, Dreger P, Pfreundschuh M, Gramatzki M, etal. Rituximab after
lymphoma-directed conditioning and allogeneic stem-cell transplantation for relapsed and
refractory aggressive non-Hodgkin lymphoma (DSHNHL R3): an open-label, randomised,
phase 2 trial. Lancet Oncol. 2014;15:757–66.
Glass B, Dohm AJ, Truemper LH, Pfreundschuh M, Bleckmann A, Wulf GG, etal. Refractory or
relapsed aggressive B-cell lymphoma failing (R)-CHOP: an analysis of patients treated on the
RICOVER-60 trial. Ann Oncol. 2017;28:3058–64.
Hutchings M, Carlo-Stella C, Bachy E, Offner FC, Morschhauser F, Crump M, etal. Glotamab
step-up dosing induces high response rates in patients with hard-to-treat refractory or relapsed
non-Hodgkin lymphoma. Blood. 2020a;136:46–8.
Hutchings M, Mous R, Clausen MR, Johnson P, Linton KM, Chamuleau MED, etal. Subcutaneous
Epcoritamab induces complete responses with an encouraging safety prole across relapsed/
refractory B-cell non-Hodgkin lymphoma subtypes, including patients with prior CAR-T ther-
apy: updated dose escalation data. Blood. 2020b;136:45–6.
van Imhoff GW, McMillan A, Matasar MJ, Radford J, Ardeshna KM, Kuliczkowski K, et al.
Ofatumumab versus rituximab salvage chemoimmunotherapy in relapsed or refractory diffuse
large B-cell lymphoma: the ORCHARRD Study. J Clin Oncol. 2017;35:544.
Kamdar M, Solomon SR, Arnason JE, Johnston PB, Glass B, Bachanova V, et al. Lisocabtagene
Maraleucel (liso-cel), a CD19-Directed Chimeric Antigen Receptor (CAR) T Cell Therapy,
Versus Standard of Care (SOC) with Salvage Chemotherapy (CT) Followed By Autologous
Stem Cell Transplantation (ASCT) As Second-Line (2L) Treatment in Patients (Pts) with
Relapsed or Refractory (R/R) Large B-Cell Lymphoma (LBCL): Results from the Randomized
Phase 3 Transform Study. Blood. 2021;138:91–91.
van Kampen RJ, Canals C, Schouten HC, Nagler A, Thomson KJ, Vernant JP, etal. Allogeneic
stem-cell transplantation as salvage therapy for patients with diffuse large B-cell non-Hodgkin’s
lymphoma relapsing after an autologous stem-cell transplantation: an analysis of the European
Group for Blood and Marrow Transplantation Registry. J Clin Oncol. 2011;29:1342–8.
Locke FL, Miklos DB, Jacobson CA, Perales MA, Kersten MJ, Oluwole OO, et al. Axicabtagene
ciloleucel as second-line therapy for large B-Cell lymphoma. N Engl J Med. 2021. 14 Dec,
Online ahead of print.
Nastoupil LJ, Jain MD, Feng L, Spiegel JY, Ghobadi A, Lin Y, etal. Standard-of-care Axicabtagene
Ciloleucel for relapsed or refractory large B-cell lymphoma: results from the US Lymphoma
CAR-T Consortium. J Clin Oncol. 2020;38:3119–28.
Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, etal. Axicabtagene
Ciloleucel CAR-T cell therapy in refractory large B-cell lymphoma. N Engl J Med.
2017;377:2531–44.
Pfreundschuh M, Kuhnt E, Trumper L, Osterborg A, Trneny M, Shepherd L, etal. CHOP-like
chemotherapy with or without rituximab in young patients with good-prognosis diffuse
large-B-cell lymphoma: 6-year results of an open-label randomised study of the MabThera
International Trial (MInT) Group. Lancet Oncol. 2011;12:1013–22.
12 Diuse Large B Cell Lymphoma andPrimary Mediastinal Lymphoma

74
Schmitz N, Nickelsen M, Ziepert M, Haenel M, Borchmann P, Schmidt C, etal. Conventional
chemotherapy (CHOEP-14) with rituximab or high-dose chemotherapy (MegaCHOEP) with
rituximab for young, high-risk patients with aggressive B-cell lymphoma: an open-label, ran-
domised, phase 3 trial (DSHNHL 2002-1). Lancet Oncol. 2012;13:1250.
Schuster SJ, Bartlett NL, Assouline S, Yoon SS, Bosch F, Sehn LH, etal. Mosunetuzumab induces
complete remissions in poor prognosis non-Hodgkin lymphoma patients, including those who
are resistant to or relapsing after chimeric antigen receptor T-cell (CAR-T) therapies, and is
active in treatment through multiple lines. Blood. 2019a;134:6.
Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, etal. Tisagenlecleucel
in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. 2019b;380:45–56.
Sehn LH, Herrera AF, Flowers CR, Kamdar MK, McMillan A, Hertzberg M, etal. Polatuzumab
vedotin in relapsed or refractory diffuse large B-cell lymphoma. J Clin Oncol. 2020a;38:155–65.
Sehn LH, Hertzberg M, Opat S, Herrera AF, Assouline SE, Flowers C, etal. Polatuzumab vedo-
tin plus bendamustine and rituximab in relapsed/refractory diffuse large B-cell lymphoma:
updated results of a phase Ib/II randomized study and preliminary results of a single-arm exten-
sion. Blood. 2020b;136:17–9.
Open Access
This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
B. Glass and M. J. Kersten

75
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_13
N. Milpied
Department of Hematology and Stem Cell Transplantation, Centre Hospitalier Universitaire,
Bordeaux, France
e-mail: [email protected]
M. Dreyling (
*)
Department of Medicine III, LMU Hospital, Munich, Germany
e-mail: [email protected]
13
Mantle Cell Lymphoma
NoelMilpied andMartinDreyling
Mantle cell lymphoma is a distinct lymphoma subtype with a widely varying clini-
cal course. Established high-risk biological factors include blastoid cytomorphol-
ogy, high cell proliferation (Ki-67>67%), and p53 mutations (Aukema etal. 2018).
While current rst-line approaches are still chemotherapy-based, BTK inhibitors
are the preferred targeted approach, especially in early relapse cases (POD24)
(Dreyling et al. 2017; Visco et al. 2021). However, cases of relapse/progression
under BTK inhibitors display extremely aggressive features with a dismal outcome
after conventional regimens (Martin etal. 2016).
Clinical Indications forCAR-T Cells
Following a conditional marketing authorization issued by the EMA in December
2020, Tecartus
®
(Gilead) is the rst autologous anti-CD-19 CAR-T cell therapy that
can be administered to patients with mantle cell lymphoma in Europe. Patients
deemed eligible for this treatment are those with histologically veried mantle cell
lymphoma resistant to or relapsing after two or more lines of treatment, including a
Bruton tyrosine kinase (BTK) inhibitor.
This registration is based on the results of recently reported a multicentre phase
2 trial (Wang etal. 2020a). Briey, 74 patients with a median age of 65 (38–79)
were enrolled, and 88% were refractory to or relapsed after BTK inhibitor treatment
at any time point. The CAR-T cell product could be manufactured for 71, and 68

76
received 2×10
6
CAR-T cells/kg on Day 0 after a conditioning regimen consisting
of udarabine (30mg/m
2
/day) and cyclophosphamide (500mg/m
2
/day) from Day 5
to Day 3. The overall response rate of all 74 patients (intent-to-treat population) was
85%, with a CR rate of 59%. More importantly, after 15months, 59% of the 60
evaluable patients were still in remission (Wang etal. 2020b) (Table13.1).
Interestingly, in contrast to conventional strategies, the percentages of patients
with an objective response were consistent among key subgroups, including patients
with high-risk features (Wang etal. 2020a).
Adverse events were mainly cytopenias (≥ grade 3: 94%) and infections (≥
grade 3: 32%). A total of 26% of the patients had grade 3 or higher cytopenias more
than 90days after the administration of KTE-X19, including neutropenia (in 16%
of patients), thrombocytopenia (16%), and anaemia (12%).
These encouraging results have also been conrmed for another CAR-T cell con-
struct (Lisocabtagene Maraleucel; Palomba etal. 2020).
Critical Evaluation
These excellent results were achieved in the context of a prospective study in highly
selected patients. Recently similar results have been reported in a “real life setting”
(Wang et al. 2021).
In the current algorithm of the approved indication, several other conditions must
be fullled before implementation of this treatment: careful work-up of the patient,
an experienced interdisciplinary team, and a specialized hospital with follow-up
resources. In future trials, the benet–risk ratio of this demanding treatment will be
rechallenged in earlier treatment lines.
Table 13.1 Updated response rates (Wang etal. 2020b)
ORR (%) CR (%)
PFS (%) OS (%)
12m 15m 12m 15m
Intent to Tx (74 patients) 85 59
Prim analysis (60 patients) 93 67 61 59 83 76
Key Points
• Patients with relapsed MCL progressing under the BTK inhibitor ibrutinib
should be considered for CD19 CAR-T cell therapy.
• Effective lymphodepleting chemotherapy is needed to allow expansion of
CAR-T cells.
N. Milpied and M. Dreyling

77
References
Aukema SM, Hoster E, Rosenwald A, etal. Expression of TP53 is associated with the outcome
of MCL independent of MIPI and Ki-67 in trials of the European MCL Network. Blood.
2018;131(4):417–20.
Dreyling M, Campo E, Hermine O, etal. Newly diagnosed and relapsed mantle cell lymphoma:
ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol.
2017;28(Suppl_4):iv62–71.
Martin P, Maddocks K, Leonard JP, etal. Postibrutinib outcomes in patients with mantle cell lym-
phoma. Blood. 2016;127(12):1559–63.
Palomba ML, Gordon LI, Siddiqi T, etal. Safety and preliminary efcacy in patients with relapsed/
refractory mantle cell lymphoma receiving Lisocabtagene Maraleucel in TRANSCEND NHL
001. ASH; 2020, #118.
Visco C, Di Rocco A, Evangelista A, et al. Outcomes in rst relapsed-refractory younger
patients with mantle cell lymphoma: results from the MANTLE-FIRST study. Leukemia.
2021;35(3):787–95.
Wang M, Munoz J, Goy A, etal. KTE-X19 CAR-T cell therapy in relapsed or refractory mantle-
cell lymphoma. N Engl J Med. 2020a;382:1331–42.
Wang M, Munoz J, Goy A, et al. One-year follow-up of ZUMA-2, the multicenter, registra-
tional study of KTE-X19 in patients with relapsed/refractory mantle cell lymphoma. ASH;
2020b, #1120.
Wang Y, Jain P, Locke FL, et al. Brexucabtagene autoleucel for relapsed/refractory mantle cell
lymphoma: real world experience from the US lymphoma CAR T consortium. ASH 2021, #744.
Open Access
This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
13 Mantle Cell Lymphoma

79
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_14
O. Tournilhac (*)
Service d’Hematologie Clinique et de Therapie Cellulaire, CHU, Universite Clermont
Auvergne, Clermont Ferrand, France
e-mail: [email protected]
P. Dreger
Department of Medicine V, Hematology, Oncology and Rheumatology, University Hospital
Heidelberg, Heidelberg, Germany
e-mail: peter[email protected]
14
Chronic Lymphocytic Leukaemia
OlivierTournilhac andPeterDreger
Clinical Development ofCAR-T Cells forCLL
Although chronic lymphocytic leukaemia (CLL) was one of the rst two entities in
which CAR-T cells were evaluated, it has not yet arrived in the clinical routine.
Since the landmark study by Porter etal. (2011), only six CLL-specic clinical tri-
als have been published, altogether comprising no more than 155 patients (Porter
etal. 2015; Gill etal. 2018; Turtle etal. 2017; Gauthier etal. 2020; Siddiqi etal.
2020; Wierda et al. 2020; Frey et al. 2020). All six of these studies investigated
CD19-directed CAR-T constructs in heavily pretreated patients, mostly having
failed BTKi +/− venetoclax therapy. Despite overall response rates of 60–95%,
including MRD clearance in a large proportion of patients, the CR rates appear to
be relatively low, and only a few durable responses have been reported in patients
achieving a CR (Porter etal. 2015; Frey etal. 2020; Cappell etal. 2020). While
toxicity includes 5–20% grade 3 cytokine release syndrome and 5–25% grade 3
neurotoxicity and appears manageable, long-term efcacy remains an unresolved
issue. CLL-specic efcacy barriers for CD19 CAR-T cells could include a reduced
capacity for sustained T cell expansion in extensively pretreated elderly CLL
patients (Lemal and Tournilhac 2019), along with impaired T cell motility, impaired
T cell mitochondrial tness, and T cell exhaustion (Bair and Porter 2019). Concurrent
use of ibrutinib might reduce the CRS rate and severity (Gauthier etal. 2020; Gill
etal. 2018; Wierda etal. 2020) without impairing CAR-T cell expansion.

80
Current Indications forCAR-T Cells intheTreatment
Landscape ofCLL
In the absence of studies with informative sample sizes and follow-up and without
an approved CAR-T cell preparation available, there is currently no indication for
CAR-T cells in CLL outside of a clinical trial. However, if a suitable trial is avail-
able, CAR-T cells can be proposed as an alternative in patients with high-risk-2
CLL who have a high transplant risk according to the EBMT-ERIC recommenda-
tions (Dreger etal. 2018). In patients with a low transplant risk, allogeneic haema-
topoietic cell transplantation (alloHCT) still appears to be the more promising
approach in terms of long-term disease control (Tournilhac etal. 2020; Roeker etal.
2020; Mato etal. 2020). The advent of more effective CAR-T cell therapies for CLL
is eagerly awaited and may rapidly change this algorithm.
Prospective Studies ofAutologous Anti-CD19 CAR-T Cell
Therapy forCLL
Porter
etal.
(2015)
Frey
etal.
(2020)
Gill
etal.
(2018)
Turtle
etal.
(2017)
Gauthier
etal.
(2020)
Siddiqi
etal.
(2020)
Wierda
etal.
(2020)
Patients (n) 14 38 19 24 (5RT) 19 (4RT) 22 (1RT) 19
CAR-T
with
ibrutinib
CTL019
No
CART-
19
No
CTL119
Yes
JCAR014
No
JCAR014
Yes
JCAR017
a
No
JCAR017
a
Yes
Age (years) 66 (51–78) 61
(49–
76)
62
(42–76)
61
(40–73)
65
(40–71)
66 (50–80) 60
(50–77)
Previous
lines (n)
5 (1–11) 3.5
(2–7)
2 (1–16) 5 (3–9) 5 (1–10) 4 (2–11) 4 (2–11)
Ibrutinib
(R/R)
1 (1) 9 (?) 5 (0) 24 (19) 19 (19) 23 (17) 19 (19)
Venetoclax
(R/R)
0 1 0 6 (6) 11 (6) 13 (11) 11 (na)
CK (%) Na Na Na 67 74 48 42
TP53 alt.
(%)
43 39
b
58 Del=58 Del=74 Mut=61
Del=35
Mut=32
Del=42
ORR (%) 57 44
b
71
b
70 83
b
82
b
95
CR (%) 29 28
b
43
b
17 22
b
46
b
63
MRD(−)
BM (%)
29 na 78
b
50
b
61
b
65
b
79
• Currently, there is no standard indication for CAR-T cells in CLL.
• CAR-T cells may be an alternative to alloHCT in high-risk patients in
clinical trials.
O. Tournilhac and P. Dreger

81
Porter
etal.
(2015)
Frey
etal.
(2020)
Gill
etal.
(2018)
Turtle
etal.
(2017)
Gauthier
etal.
(2020)
Siddiqi
etal.
(2020)
Wierda
etal.
(2020)
CRS (all/
G3) (%)
64/43 63/24 95/16 83/8 74/0 74/9 74/5
NT (all/
G3) (%)
36/7 na/8 26/5 33/25 26/26 39/22 32/16
FU (m) 19 (6–53) 32
(2–75)
19
(8–28)
NA 12 (4–17) 24 10
PFS (m)
PFS >24m
(n)
28%
@18m
3
1m
7
na
na
8.5m
na
38%
@12m
na
50%
@18m
na
na
na
NRM (n)
cause
1
(infection)
0
/
1
(cardiac)
1
(CRS/
NT)
1
(cardiac)
0
/
0
/
RT Richter transformation, R/R relapsed/refractory, TP53 alt. TP53 mutation and/or 17p deletion,
CRS cytokine release syndrome, NT neurotoxicity, allG all grades, G≥3 grade≥3, na not avail-
able, CK complex karyotype (≥3 abnormalities), BM bone marrow, MRD(−) BM negative bone
marrow minimal residual disease, NRM non relapse mortality
a
Transcend CLL 004 study with lisocabtagene maraleucel
b
Assessment limited to evaluable patients
References
Bair SM, Porter DL.Accelerating chimeric antigen receptor therapy in chronic lymphocytic leu-
kemia: the development and challenges of chimeric antigen receptor T-cell therapy for chronic
lymphocytic leukemia. Am J Hematol. 2019;94(Suppl_1):S10–7.
Cappell KM, Sherry RM, Yang JC, et al. Long-term follow-up of anti-CD19 chimeric antigen
receptor T-cell therapy. JCO. 2020;38(32):3805–15.
Dreger P, Ghia P, Schetelig J, etal. High-risk chronic lymphocytic leukemia in the era of pathway
inhibitors: integrating molecular and cellular therapies. Blood. 2018;132(9):892–902.
Frey NV, Gill S, Hexner EO, et al. Long-term outcomes from a randomized dose optimization
study of chimeric antigen receptor modied T cells in relapsed chronic lymphocytic leukemia.
JCO. 2020;38:2862.
Gauthier J, Hirayama AV, Purushe J, etal. Feasibility and efcacy of CD19-targeted CAR-T cells
with concurrent ibrutinib for CLL after ibrutinib failure. Blood. 2020;135(19):1650–60.
Gill SI, Vides V, Frey NV, etal. Prospective clinical trial of anti-CD19 CAR-T cells in combination
with Ibrutinib for the treatment of chronic lymphocytic leukemia shows a high response rate.
Blood. 2018;132(Suppl 1):298.
Key Points
• Autologous CAR-T cells for CLL have been in development for almost
10years, with interesting results in poor-risk disease, including patients
double refractory to both BTKi and BCL2i.
• However, more data, including clinical trials with a longer follow-up time,
are required before adding CAR-T cells to clinical practice.
14 Chronic Lymphocytic Leukaemia

82
Lemal R, Tournilhac O.State-of-the-art for CAR-T cell therapy for chronic lymphocytic leukemia
in 2019. J Immunother Cancer. 2019;7(1):202.
Mato AR, Roeker LE, Jacobs R, etal. Assessment of the efcacy of therapies following Venetoclax
discontinuation in CLL reveals BTK inhibition as an effective strategy. Clin Cancer Res.
2020;26(14):3589–96.
Porter DL, Levine BL, Kalos M, Bagg A, June CH.Chimeric antigen receptor–modied T cells in
chronic lymphoid leukemia. N Engl J Med. 2011;365(8):725–33.
Porter DL, Hwang W-T, Frey NV, et al. Chimeric antigen receptor T cells persist and induce
sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med.
2015;7(303):303ra139.
Roeker LE, Dreger P, Brown JR, etal. Allogeneic stem cell transplantation for chronic lympho-
cytic leukemia in the era of novel agents. Blood Adv. 2020;4(16):3977–89.
Siddiqi T, Soumerai JD, Dorritie KA, etal. Updated follow-up of patients with relapsed/refrac-
tory chronic lymphocytic leukemia/small lymphocytic lymphoma treated with Lisocabtagene
Maraleucel in the phase 1 monotherapy cohort of transcend CLL 004, including high-risk and
Ibrutinib-treated patients. Blood 2020;136(1):546.
Tournilhac O, Le Garff-Tavernier M, Nguyen Quoc S, etal. Efcacy of minimal residual disease
driven immune-intervention after allogeneic hematopoietic stem cell transplantation for high-
risk chronic lymphocytic leukemia: results of a prospective multicentric trial. Haematologica.
2020; https://doi.org/10.3324/haematol.2019.239566.
Turtle CJ, Hay KA, Hana L-A, etal. Durable molecular remissions in chronic lymphocytic leu-
kemia treated with CD19-specic chimeric antigen receptor-modied T cells after failure of
Ibrutinib. J Clin Oncol. 2017;35(26):3010–20.
Wierda W, Dorritie KA, Munoz J, et al. Transcend CLL 004: phase 1 Cohort of Lisocabtagene
Maraleucel (liso-cel) in combination with Ibrutinib for patients with Relapsed/Refractory
(R/R) chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL). Blood
2020;136(1):544.
Open Access
This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
O. Tournilhac and P. Dreger

83
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_15
F. Morschhauser (*)
Department of Hematology, University of Lille, CHU Lille, ULR 7365– GRITA– Groupe de
Recherche sue les forms injectables et les Technologies Associees, Lille, France
e-mail: [email protected]
P. L. Zinzani
Institute of Hematology “L. e A.Seràgnoli” University of Bologna, Bologna, Italy
e-mail: [email protected]
15
Indolent Lymphomas
FranckMorschhauser andPierLuigiZinzani
Indolent non-Hodgkin lymphoma (iNHL), including follicular (FL) and marginal
zone (MZL) lymphoma, now enjoy durable disease control with rst-line immuno-
chemotherapy, with a median overall survival (OS) of over 15years in most series
(Kahl and Yang 2016). However, iNHL is still widely considered incurable in most
cases, and disease history remains characterized by a relapsing and remitting course,
with each remission period shorter than the previous one, and OS and progression-
free survival (PFS) decrease with each subsequent line of conventional therapy
(Batlevi etal. 2020). Patients with unmet needs include approximately 20% of FL
patients who experience disease progression within 24months (POD24) after initial
chemoimmunotherapy (with a 5-year OS of 48% (Casulo etal. 2015)—although it
remains unclear how much this worse outcome is driven by misdiagnosed trans-
formed follicular lymphoma (Freeman etal. 2019)); those who fail multiple regi-
mens (5-year PFS of 23%) (Rivas-Delgado et al. 2019), have double refractory
disease (Gopal etal. 2017) or experience relapse after autologous stem cell trans-
plantation (ASCT) (Sesques etal. 2020). Although promising results were obtained
with an immunomodulatory regimen combining anti-CD20 Moab and lenalidomide
(Leonard etal. 2019; Morschhauser etal. 2019), most current approved therapies do
not overcome incremental disease resistance, resulting in multiple lines of treatment
with cumulative toxicity over a patient’s lifetime. The autologous anti-CD19 chime-
ric antigen receptor T cell (CAR-T) therapies tisa-cel and axi-cel, which are now
approved for patients with relapsed/refractory (r/r) large B cell lymphoma (LBCL),
have also been tested in iNHL, with promising results.

84
The ZUMA-5 phase 2 trial evaluated the efcacy and safety of axi-cel in 146
patients with r/r iNHL (FL: 124; MZL: 22) after at least two lines of therapy
(Jacobson etal. 2020). Among the 104 patients available for the efcacy analysis
(84 with FL and 20 with MZL), the overall response rate (ORR) was 92%, with 76%
of patients obtaining complete remission (CR). In FL patients, the ORR was 94%,
with a CR rate of 80%. Response rates were consistent among patients with high-
risk features. With a median follow-up of 17.5months, 64% of FL patients remained
in response. The median duration of response (DoR), PFS, and OS were not reached
(Gopal etal. 2017). The safety prole was manageable and appeared favourable in
patients with FL compared with that previously reported in LBCL (Neelapu etal.
2017; Locke etal. 2019). Grade≥3 adverse events (AEs) occurred in 126 patients
(86%), most commonly neutropenia and infection. Fewer instances of any grade
(78%) and high-grade (6%) cytokine release syndrome (CRS) were observed in the
FL cohort. The onset of CRS was delayed compared with that seen in LBCL.The
event was not resolved in only one patient, who ultimately died due to multiorgan
failure (Jacobson etal. 2020). Fifty-six percent of patients experienced neurological
events (NEs) of any grade; 15% had grade≥3 events. Most NEs (67/70) resolved
by the data cut-off time (Jacobson etal. 2020).
The same reliable results were seen with tisa-cel. In the phase 2 ELARA study,
98 adult patients with r/r FL within 6months after second or later therapy or that
relapsed after ASCT were enrolled (Fowler et al. 2020). Ninety-seven patients
received tisa-cel, but 52 were evaluable for efcacy. Unlike the ZUMA-5 trial,
bridging therapy was allowed, and 43% of patients received it. Thirty-four of 52
patients (65.4%) achieved a CR, with an ORR of 82.7%. With a median follow-up
of 9.9months, 69% of patients were still in response. Median DoR, PFS, and OS
were not reached. Of the 97 patients evaluable for safety (median follow-up of
6.6months), 69% experienced grade≥3 AEs, most commonly neutropenia; 48% of
patients had CRS, but none of them experienced a grade≥3 AE.Any grade NEs
occurred in 10% of patients; 2% had a grade≥3 NE, and all recovered. No deaths
seen were treatment-related (Fowler etal. 2020).
These preliminary data from the ELARA and ZUMA-5 trials suggest that anti-
CD19 CAR-T cell treatment is effective in high-risk or extensively treated r/r iNHL,
resulting in a high CR and ORR. Although the benet/risk ratio seems highly
favourable in high-risk FL patients, such as young, double refractory, relapse post-
ASCT patients or those with POD24, longer follow-up times are needed to better
dene the potential for cure and the limited long-term toxicities, especially in view
of the emergence of highly efcient competitive therapies, such as bispecic anti-
bodies (Bannerji etal. 2020; Hutchings etal. 2020a,b; Assouline etal. 2020). Data
remain scarce in MZL.Clearly, phase III randomized trials are mandatory to con-
rm the role of CAR-T cells in R/R in NHL, especially in POD24 patients.
F. Morschhauser and P. L. Zinzani

85
References
Assouline SE, Kim WS, Sehn LH, etal. Mosunetuzumab shows promising efcacy in patients
with multiply relapsed follicular lymphoma: updated clinical experience from a phase I dose-
escalation trial. ASH Congress; 2020, abstract 702.
Bannerji R, Allan JN, Arnason JE, et al. Odronextamab (REGN1979), a human CD20 x CD3
bispecic antibody, induces durable, complete responses in patients with highly refrac-
tory B-cell non-Hodgkin lymphoma, including patients refractory to CAR-T therapy ASH
Congress; 2020, abstract 400.
Batlevi L, Sha F, Alperovich A, et al. Blood. Cancer J. 2020;10(7):74. https://doi.org/10.1038/
s41408- 020- 00340- z.
Casulo C, Byrtek M, Dawson KL, etal. Early relapse of follicular lymphoma after rituximab plus
cyclophosphamide, doxorubicin, vincristine, and prednisone denes patients at high risk for
death: an analysis from the National LymphoCare Study. J Clin Oncol. 2015;33(23):2516–22.
https://doi.org/10.1200/JCO.2014.59.7534. Epub 2015 Jun 29.
Fowler NH, Dickinson M, Dreyling M, et al. Efcacy and safety of tisagenlecleucel in adult
patients with relapsed/refractory follicular lymphoma: interim analysis of the phase 2 Elara
Trial ASH Congress; 2020, abstract 1149.
Freeman CL, Kridel R, Moccia AA, et al. Early progression after bendamustine-rituximab is
associated with high risk of transformation in advanced stage follicular lymphoma. Blood.
2019;134(9):761–4. https://doi.org/10.1182/blood.2019000258. Epub 2019 Jul 12.
Gopal AK, Kahl BS, Flowers CR, etal. Idelalisib is effective in patients with high-risk follicular
lymphoma and early relapse after initial chemoimmunotherapy. Blood. 2017;129(22):3037–9.
https://doi.org/10.1182/blood- 2016- 12- 757740. Epub 2017 Mar 21.
Hutchings M, Mous R, Roost Clausen M, et al. Subcutaneous Epcoritamab induces complete
responses with an encouraging safety prole across relapsed/refractory B-cell non-Hodgkin
lymphoma subtypes, including patients with prior CAR-T therapy: updated dose escalation
data. ASH Congress 2020a, abstract 402.
Hutchings M, Carlo-Stella C, Bachy E, etal. Glotamab step-up dosing induces high response
rates in patients with hard-to-treat refractory or relapsed non-Hodgkin lymphoma. ASH
Congress; 2020b, abstract 403.
Jacobson C, Chavez JC, Sehgal AR, et al. Primary analysis of Zuma-5: a phase 2 study of
Axicabtagene Ciloleucel (Axi-Cel) in patients with relapsed/refractory (R/R) indolent non-
Hodgkin lymphoma (iNHL). ASH Congress; 2020, abstract700.
Kahl BS, Yang DT. Follicular lymphoma: evolving therapeutic strategies. Blood.
2016;127(17):2055–63. https://doi.org/10.1182/blood- 2015- 11- 624288.
Key Points
• Anti-CD19 CAR-T cell treatment achieves high CR and ORRs in exten-
sively treated r/r FLs with an acceptable safety prole.
• The response appears durable, but the median follow-up time remains short.
• Data remain scarce in MZL.
• Phase 3 randomized trials are mandatory to conrm the role of CAR-T
cells in r/r iNHL, especially in POD24 patients.
15 Indolent Lymphomas

86
Leonard JP, Trneny M, Izutsu K, etal. AUGMENT: a phase III study of Lenalidomide plus ritux-
imab versus placebo plus rituximab in relapsed or refractory indolent lymphoma. J Clin Oncol.
2019;37(14):1188–99. https://doi.org/10.1200/JCO.19.00010. Epub 2019 Mar 21.
Locke FL, Ghobadi A, Jacobson CA, etal. Long-term safety and activity of axicabtagene cilo-
leucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1-2
trial. Lancet Oncol. 2019;20(1):31–42. https://doi.org/10.1016/S1470- 2045(18)30864- 7. Epub
2018 Dec 2.
Morschhauser F, Le Gouill S, Feugier P, etal. Obinutuzumab combined with lenalidomide for
relapsed or refractory follicular B-cell lymphoma (GALEN): a multicentre, single-arm, phase 2
study. Lancet Haematol. 2019;6(8):e429–37. https://doi.org/10.1016/S2352- 3026(19)30089- 4.
Epub 2019 Jul 8.
Neelapu SS, Locke FL, Bartlett NL, etal. Axicabtagene Ciloleucel CAR-T cell therapy in refrac-
tory large B-cell lymphoma. N Engl J Med. 2017;377(26):2531–44. https://doi.org/10.1056/
NEJMoa1707447. Epub 2017 Dec 10.
Rivas-Delgado A, Laura Magnano L, Moreno-Velázquez M, etal. Response duration and survival
shorten after each relapse in patients with follicular lymphoma treated in the rituximab era. Br
J Haematol. 2019;184(5):753–9. https://doi.org/10.1111/bjh.15708. Epub 2018 Dec 4.
Sesques P, Bourcier J, Goler C, etal. Clinical characteristics and outcomes of relapsed follicular
lymphoma after autologous stem cell transplantation in the rituximab era. Hematol Oncol.
2020;38(2):137–45. https://doi.org/10.1002/hon.2713. Epub 2020 Jan 30.
Open Access
This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
F. Morschhauser and P. L. Zinzani

87
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_16
I. Yakoub-Agha
Maladies du Sang, Unité de Thérapie Cellulaire, Centre hospitalier-Universitaire de Lille,
Lille, France
e-mail: [email protected]
H. Einsele (
*)
Department of Internal Medicine II, University Hospital Würzburg, Würzburg,
Bayern, Germany
e-mail: Einsele_H@ukw.de
16
Multiple Myeloma
IbrahimYakoub-Agha
andHermannEinsele
To date, over 100 clinical trials investigating the use of CAR-T cells in MM have
been registered at clinicaltrials.gov. Although several CD19-directed CAR-T cell
products have been approved (Ghobadi 2018; Yassine etal. 2020), CD19 surface
expression on plasma cells is limited or absent, leading to uncertain efcacy in clini-
cal trials that used anti-CD19 alone in patients with MM (Garfall etal. 2015, 2019).
Using superresolution microscopy, CD19 can be detected on a large proportion of
myeloma cells, which could explain the successful targeting and lysis of myeloma
cells by CD19-detecting CAR-T cells (Nerreter etal. 2019). Of note, some ongoing
studies in which CD19 is targeted in combination with other antigens, especially
BCMA, are being conducted (Beauvais etal. 2020).
BCMA-directed CAR-T cells have shown promising efcacy and safety proles
in various phase I/II clinical trials (Munshi etal. 2021; Brudno etal. 2018; Cohen
etal. 2019; Mailankody etal. 2020; Zhao etal. 2018). Indeed, the overall response
rate ranged from 75 to 100%, with median event-free survival ranging from 11 to
24months, in heavily pretreated MM patients, which is far better than all other cur-
rently available drugs and agents in this patient cohort.
At least two BCMA-directed CAR-T cell products will likely move into routine
clinical use in the near future. Outside clinical trials, the main indication for CAR-T
cell therapy in MM would be limited to patients with R/R MM after at least two
lines of prior therapy that included PIs (proteasome inhibitors), iMIDs

88
(immunomodulatory agents, e.g., lenalidomide, pomalidomide), and anti-C38
monoclonal antibodies.
Although CAR-T cell therapy appears promising, the duration of disease control
is limited, and almost all patients ultimately relapse. This might partially reect the
fact that CAR-T cells have thus far only been given to heavily pretreated patients
with advanced, resistant disease (Beauvais etal. 2020; Gauthier and Yakoub-Agha
2017). Thus, CAR-T cell exhaustion and the reduction and even irreversible loss of
expression of the target antigen BCMA on tumour cells (Da Vià etal. 2021; Samur
etal. 2021), which are often genetically highly unstable, are other factors limiting
CAR-T cell efcacy. Thus, novel targets or even dual targeting (including targeting
of GPRC5D (de Larrea et al. 2020), SLAMF7 (Gogishvili et al. 2017), CD229
(Radhakrishnan etal. 2020), and CD38 (Gauthier and Yakoub-Agha 2017; Verkleij
etal. 2020)) is currently being explored.
Additionally, to increase efcacy, CAR-T cell therapy is moved to earlier lines of
therapy to increase the tness and persistence of the generated MM-specic CAR-T
cells. However, CAR-T cell therapy in MM—less when compared to patients with
aggressive lymphomas—can be associated with substantial, potentially life-
threatening toxicity. Thus, administering CAR-T cells with a better effector func-
tion and proliferation potential may be a challenge as CAR-T therapy is used at
earlier stages of disease (Prommersberger etal. 2018).
References
Beauvais D, Danhof S, Hayden PJ, Einsele H, Yakoub-Agha I.Clinical data, limitations and per-
spectives on chimeric antigen receptor T-cell therapy in multiple myeloma. Curr Opin Oncol.
2020;32(5):418–26.
Brudno JN, Maric I, Hartman SD, Rose JJ, Wang M, Lam N, Stetler-Stevenson M, Salem D, Yuan
C, Pavletic S, etal. T cells genetically modied to express an anti-B-cell maturation antigen
chimeric antigen receptor cause remissions of poor-prognosis relapsed multiple myeloma. J
Clin Oncol. 2018;36(22):2267–80.
Cohen AD, Garfall AL, Stadtmauer EA, Melenhorst JJ, Lacey SF, Lancaster E, Vogl DT, Weiss
BM, Dengel K, Nelson A, etal. B cell maturation antigen-specic CAR-T cells are clinically
active in multiple myeloma. J Clin Invest. 2019;129(6):2210–21.
Key Points
• BCMA is the major target for CAR-T cell therapy in MM.
• Two BCMA-directed CAR-T cell products are moving into the clinical
routine in the near future (one has already been approved by the FDA
and EMA).
• Irreversible BCMA loss on MM cells has been described in a few patients
as a cause of failure of BCMA-directed CAR-T cells.
• CAR-T cell therapy is moving to earlier lines of therapy in MM patients.
I. Yakoub-Agha and H. Einsele

89
Da Vià MC, Dietrich O, Truger M, Arampatzi P, Duell J, Heidemeier A, Zhou X, Danhof S,
Kraus S, Chatterjee M, Meggendorfer M, Twardziok S, Goebeler ME, Topp MS, Hudecek M,
Prommersberger S, Hege K, Kaiser S, Fuhr V, Weinhold N, Rosenwald A, Erhard F, Haferlach
C, Einsele H, Kortüm KM, Saliba AE, Rasche L.Homozygous BCMA gene deletion in response
to anti-BCMA CAR-T cells in a patient with multiple myeloma. Nat Med. 2021;27(4):616–19.
Garfall AL, Maus MV, Hwang WT, Lacey SF, Mahnke YD, Melenhorst JJ, Zheng Z, Vogl DT,
Cohen AD, Weiss BM, et al. Chimeric antigen receptor T cells against CD19 for multiple
myeloma. N Engl J Med. 2015;373(11):1040–7.
Garfall AL, Stadtmauer EA, Hwang WT, Lacey SF, Melenhorst JJ, Krevvata M, Carroll MP,
Matsui WH, Wang Q, Dhodapkar MV et al. Anti-CD19 CAR-T cells with high-dose mel-
phalan and autologous stem cell transplantation for refractory multiple myeloma. JCI Insight.
2019;4(4):e127684.
Gauthier J, Yakoub-Agha I. Chimeric antigen-receptor T-cell therapy for hematological malig-
nancies and solid tumors: clinical data to date, current limitations and perspectives. Curr Res
Transl Med. 2017;65(3):93–102.
Ghobadi A.Chimeric antigen receptor T cell therapy for non-Hodgkin lymphoma. Curr Res Transl
Med. 2018;66(2):43–9.
Gogishvili T, Danhof S, Prommersberger S, Rydzek J, Schreder M, Brede C, Einsele H, Hudecek
M.SLAMF7-CAR-T cells eliminate myeloma and confer selective fratricide of SLAMF7(+)
normal lymphocytes. Blood. 2017;130(26):2838–47.
de Larrea CF, Staehr M, Lopez AV, Ng KY, Chen Y, Godfrey WD, Purdon TJ, Ponomarev V,
Wendel HG, Brentjens RJ, Smith EL.Dening an optimal dual-targeted CAR-T cell therapy
approach simultaneously targeting BCMA and GPRC5D to prevent BCMA escape-driven
relapse in multiple myeloma. Blood Cancer Discov. 2020;1(2):146–54.
Mailankody S, Jakubowiak AJ, Htut M, Costa LJ, Lee K, Ganguly S, Kaufman JL, Siegel DSD,
Bensinger W, Cota M, et al. Orvacabtagene autoleucel (orva-cel), a B-cell maturation anti-
gen (BCMA)-directed CAR-T cell therapy for patients (pts) with relapsed/refractory multiple
myeloma (RRMM): update of the phase 1/2 EVOLVE study (NCT03430011). J Clin Oncol.
2020;38(15_Suppl):8504.
Munshi NC, Anderson LD Jr, Shah N, Madduri D, Berdeja J, Lonial S, Raje N, Lin Y, Siegel D,
Oriol A, etal. Idecabtagene Vicleucel in relapsed and refractory multiple myeloma. N Engl J
Med. 2021;384(8):705–16.
Nerreter T, Letschert S, Götz R, Doose S, Danhof S, Einsele H, Sauer M, Hudecek M. Super-
resolution microscopy reveals ultra-low CD19 expression on myeloma cells that triggers elimi-
nation by CD19 CAR-T.Nat Commun. 2019;10(1):3137.
Prommersberger S, Jetani H, Danhof S, Monjezi R, Nerreter T, Beckmann J, Einsele H, Hudecek
M.Novel targets and technologies for CAR-T cells in multiple myeloma and acute myeloid
leukemia. Curr Res Transl Med. 2018;66(2):37–8.
Radhakrishnan SV, Luetkens T, Scherer SD, Davis P, Vander Mause ER, Olson ML, Yousef S,
Panse J, Abdiche Y, Li KD, Miles RR, Matsui W, Welm AL, Atanackovic D.CD229 CAR-T
cells eliminate multiple myeloma and tumor propagating cells without fratricide. Nat Commun.
2020;11(1):798.
Samur MK, Fulciniti M, Aktas Samur A, Bazarbachi AH, Tai YT, Prabhala R, Alonso A,
Sperling AS, Campbell T, Petrocca F, Hege K, Kaiser S, Loiseau HA, Anderson KC, Munshi
NC.Biallelic loss of BCMA as a resistance mechanism to CAR-T cell therapy in a patient with
multiple myeloma. Nat Commun. 2021;12(1):868.
Verkleij CPM, Jhatakia A, Broekmans MEC, Frerichs KA, Zweegman S, Mutis T, Bezman NA,
van de Donk NWCJ.Preclinical rationale for targeting the PD-1/PD-L1 Axis in combination
with a CD38 antibody in multiple myeloma and other CD38-positive malignancies. Cancers
(Basel). 2020;12(12):3713.
Yassine F, Iqbal M, Murthy H, Kharfan-Dabaja MA, Chavez JC. Real world experience of
approved chimeric antigen receptor T-cell therapies outside of clinical trials. Curr Res Transl
Med. 2020;68(4):159–70.
16 Multiple Myeloma

90
Zhao WH, Liu J, Wang BY, Chen YX, Cao XM, Yang Y, Zhang YL, Wang FX, Zhang PY, Lei B,
etal. A phase 1, open-label study of LCAR-B38M, a chimeric antigen receptor T cell therapy
directed against B cell maturation antigen, in patients with relapsed or refractory multiple
myeloma. J Hematol Oncol. 2018;11(1):141.
Open Access
This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
I. Yakoub-Agha and H. Einsele

91
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_17
P. Corradini
Divisione di Ematologia, Fondazione IRCCS Istituto Nazionale dei Tumori di Milano,
Università degli Studi di Milano, Milan, Italy
e-mail: [email protected]
L. Trümper (
*)
Department of Hematology and Oncology, Georg-August University Göttingen,
Göttingen, Germany
e-mail: [email protected]
17
Developments inOther Haematological
Malignancies: Other Lymphoid
Malignancies
PaoloCorradini andLorenzTrümper
Peripheral T cell lymphomas comprise a heterogeneous group of rare diseases, rep-
resenting 10–15% of all non-Hodgkin lymphomas (NHLs). Upfront treatment for
peripheral T cell lymphoma (pTNHL) includes CHOP-like (cyclophosphamide,
adriamycin, vincristine, prednisone) multiagent chemotherapy with or without eto-
poside, followed by stem cell transplantation as consolidation in responsive t
patients. This approach induces durable long-term remission in approximately 40%
of cases; early refractoriness during induction occurs in approximately 25% of
patients, with the remaining patients typically relapsing within 24months. With the
exception of patients with anaplastic large cell lymphomas who are eligible to
receive brentuximab vedotin, there is no standard of care in the relapse setting. In
patients not eligible to receive high-dose chemotherapy followed by allogeneic stem
cell transplantation, the prognosis is dismal.
CAR-T cells have shown impressive results in relapsed/refractory B-cell lym-
phoma and are currently under investigation in T cell lymphomas.
Target Antigens
The choice of the appropriate antigen constitutes the main challenge in targeting T
cell malignancies using CAR-T cells. Many target antigens are expressed by both
physiological T cells and engineered CAR-T cells (Tables 17.1 and 17.2).

92
Therefore, this shared antigen expression can potentially result in the follow-
ing issues:
– A fratricide effect on CAR-T cells.
– Ablation of physiological donor T cells after CAR-T cell infusion, leading to
deep and/or long-lasting immune deciency and T cell aplasia.
CAR-T Development inT Cell Malignancies
Some experimentally engineered CAR-T cell products targeting CD5, CD7, CD30,
and TRBC1 (T cell receptor beta chain 1) have been tested (Table17.3).
Table 17.1 Pan-T cell antigens
CD5 Expression in T cells, thymocytes, B-1 cells, and T cell malignancies:
90% T-ALL/Ly
85% PTCL-nos
96% AITL
26–32% ALCL
36% NK-T
85% ATLL
91% CTCL
CD7 Expression in T cells, thymocytes, NK cells, and T cell malignancies:
95% T-ALL/Ly
50% PTCL-nos
57% AITL
32–54% ALCL
79% NK-T
25% ATLL
18% CTCL
Table 17.2 Antigens with restricted expression
CD30 Expression in activated T and B cells and in T cell malignancies:
17% T-ALL/Ly
16% PTCL-nos
32–50% AITL
93% ALCL
64% NK-T
39% ATLL
18% CTCL
TRBC1 Expression in T cells and in T cell malignancies:
7–11% T-ALL/Ly
27% PTCL-nos
34% AITL
25% ALCL
P. Corradini and L. Trümper

93
Table 17.3 CAR-T cells targeting T lymphocyte antigens
CAR-T cells
targeting
CD5
Mamonkin etal. (Blood 2015), preclinical experience:
– CD5 CAR-T cells eliminate malignant T-ALL/Ly lines invitro and inhibit
disease progression in xenograft mouse models
– Second-generation CD5 CAR with a CD28 costimulatory domain: With the
loss of CD5 expression on the surface of T cells, CD5 CAR-T cells become
resistant to fratricide
Hill etal. (Blood 2019), phase I dose escalation study, MAGENTA trial:
– 9 patients enrolled (4T-ALL, 5T-NHL)
– CD5 CAR-T cells are safe and can induce clinical responses (3 patients in
complete response) in heavily pretreated relapsed/refractory T-ALL and
T-NHL, without inducing T cell aplasia
CAR-T cells
targeting
CD7
Gomes-Silva etal. (Blood 2017), preclinical models of T cell malignancies:
– Fulminant fratricide precluding expansion of CAR-T cells
– Abrogation of CD7 expression from the cell surface shows potential activity
A phase I study (CRIMSON trial) has been designed at Baylor College of
Medicine but is not yet recruiting
CAR-T cells
targeting
TRBC1
Maciocia etal. (Nat Med 2017), preclinical studies:
– CAR-T cells targeting TRBC1 are able to specically eliminate malignant T
cell lines expressing TRBC1
– TRBC1 CAR-T cells cannot target normal TRBC2-positive T cells
A phase I/II study (AUTO4) coordinated by the University College of London
is a single-arm trial evaluating the safety and clinical activity of a CAR-T cell
targeting TRBC1in patients with relapsed/refractory TRBC1-positive T cell
lymphomas
CAR-T cells
targeting
CD30
Dotti etal. (Immunol Rev. 2014), preclinical studies:
– CAR-T cells targeting CD30 generate tumour-specic T cells in patients with
Hodgkin and anaplastic T cell lymphomas
– Tumour recognition by CD30 CAR-T cells is MHC-unrestricted
– CAR-T cells targeting CD30 potentially overcome tumour escape
Several small clinical trials are being reported; some studies are ongoing and
recruiting:
– Two CAR-T constructs are under investigation, one CAR-T cell with the
antigen-binding domain of the anti-CD30 and ant-CD28 costimulatory
domain and another CAR-T cell targeting CD30 and 4-1BB as a
costimulatory domain
– In the Ramos etal. phase I study, 9 patients with relapsed/refractory Hodgkin
and EBV-negative, CD30-positive ALCL have been treated; results are
promising, with 1 patient in complete remission and 3in stable disease,
without relevant toxicities
– Wang etal. enrolled 18 patients (17 Hodgkin, one ALCL); seven patients
achieved a partial response and six achieved stable disease, with limited
acute toxicities but an increased risk of infections
– Grover etal. enrolled 24 patients (Hodgkin, ALCL, EATL, and Sezary
syndrome) in a phase Ib/II study with anti-CD30 CAR-T cells, which
demonstrated early clinical effects and good tolerability and safety
– A phase I study is ongoing at the National Cancer Institute to assess safety
and feasibility in advanced CD30-positive ALCL and PTCL-NOS
17 Developments in Other Haematological Malignancies: Other Lymphoid…

94
Bibliography
Dotti G, Gottschalk S, Savoldo B, etal. Design and development of therapies using chimeric anti-
gen receptor-expressing T cells. Immunol Rev. 2014;257(1):107–26.
Foster C, Kuruvilla J.Treatment approaches in relapsed or refractory peripheral T-cell lymphomas.
F1000Research. 2020;9(Faculty Rev):1091.
Gomes-Silva D, Srinivasan M, Sharma S, etal. CD7-edited T cells expressing a CD7-specic CAR
for the therapy of T-cell malignancies. Blood. 2017;130:285–96.
Grover NS, Park SI, Ivanova A, etal. A phase Ib/II study of anti-CD30 chimeric antigen recep-
tor T cells for relapsed/refractory CD30+ lymphomas. Biol Blood Marrow Transplant.
2019;25(3):S66.
Hill LC, Rouce RH, Smith TS, etal. Safety and anti-tumor activity of CD5 CAR-T cells in patients
with relapsed/refractory T-cell malignancies. Blood. 2019;134(S1):199.
Maciocia PM, Wawrzyniecka PA, Philip B, etal. Targeting the T cell receptor β-chain constant
region for immunotherapy of T cell malignancies. Nat Med. 2017;23(12):1416–23.
Mamonkin M, Rouce RH, Tashiro H, et al. A T-cell directed chimeric antigen receptor for the
selective treatment of T-cell malignancies. Blood. 2015;126:983–92.
Ramos CA, Ballard B, Zhang H, etal. Clinical and immunological responses after CD30-specic
chimeric antigen receptor–redirected lymphocytes. J Clin Invest. 2017;127(9):3462–71.
Rogers AM, Brammer JE.Hematopoietic cell transplantation and adoptive cell therapy in periph-
eral T cell lymphoma. Curr Hematol Malig Rep. 2020;15:316–32.
Sherer LD, Brenner MK, Mamonkin M.Chimeric antigen receptors for T-cell malignancies. Front
Oncol. 2019;9(126):1–10.
Vose J, Armitage J, Weisenburger D.International peripheral T-cell and natural killer/T-cell lym-
phoma study: pathology ndings and clinical outcomes. J Clin Oncol. 2008;26(25):4124–30.
Wang CM, Wu ZQ, Wang Y, etal. Autologous T cells expressing CD30 chimeric antigen receptors
for relapsed or refractory Hodgkin lymphoma: an open-label phase I trial. Clin Cancer Res.
2017;23(5):1156–66.
Key Points
• Target antigens are expressed by normal T cells, malignant T cells, and
engineered CAR-T cells.
• Therefore, the major concern for targeting T cell malignancies with CAR-T
cells is a fratricide effect.
• A second major issue is the ablation of normal T cells after CAR-T cell
infusion, potentially causing severe and/or long-lasting immune deciency
and T cell aplasia.
• Currently, the most promising constructs are CAR-T cells targeting CD30.
• Phase I and II studies are ongoing in T cell malignancies and Hodgkin
lymphoma, thus far demonstrating feasibility, tolerability, and potential for
clinical efcacy.
P. Corradini and L. Trümper

95
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
17 Developments in Other Haematological Malignancies: Other Lymphoid…

97
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_18
C. Ferrand
EFS– INSERM UMR1098 RIGHT– UBFC, Molecular Onco Hematology Lab, EFS
Bourgogne Franche-Comté, Besançon, France
e-mail: [email protected]
A. Rambaldi (
*)
Department of Oncology-Hematology, University of Milan and Azienda Socio Sanitaria
Territoriale Papa Giovanni XXIII, Bergamo, Italy
e-mail: [email protected]
18
Myeloid Malignancies
ChristopheFerrand andAlessandroRambaldi
In addition to chemotherapy, which remains the basic treatment, the treatment panel
for acute myeloid leukaemia (AML) has expanded considerably in recent years.
Clinicians now have a large choice of therapies: targeted therapies (anti-IDH1/2,
anti-FLT3, and anti-BCL2 therapies, among others), drugs targeting epigenetic
mechanisms, kinase inhibitors (FLT3, MAPK, and JAK2, etc.), immunotherapies
(monoclonal antibodies linked or not to a toxin, dual/bispecic), and cellular immu-
notherapies. Moreover, despite its toxicities, allogeneic transplantation often
remains an effective nal therapeutic alternative. However, most patients are refrac-
tory or relapsed (R/R) after several lines of therapy. Thus, there is a clinical need in
AML R/R patients, and CAR-T cells may be an option and can nd a place in the
treatment to reduce tumour burden and clinical evolution of the disease (Fig.18.1,
modied from Roussel etal. (2020)).
Several currently ongoing research programs aim to generate CAR-T cells
against myeloid malignancies (Hofmann etal. 2019). However, the absence of a
truly AML-specic marker generates remarkable uncertainty regarding the optimal
antigens to target, and signicant concern remains about off-target effects on nor-
mal haematopoiesis. The difculty of obtaining successful manufacture of CAR-T
cells from heavily pretreated patients has paved the way to investigation of different
cell sources to build alternative platforms for cellular therapy.

98
Single or Dual Antigen Targeting?
CAR-T cells targeting CD33 and CD123 have already been investigated in early
phase clinical trials. Unfortunately, these antigens do not avoid “on-target off-
tumour” effects, such as myelotoxicity and endothelial toxicity. For this reason,
CAR-T cells directed against other surface proteins, such as CCL-1, CD44v6,
FLT3, c-KIT (CD117), CD38, B7-H3 (also known as CD276), NKG2D, and
IL-1RAP, are also under preclinical and clinical investigation (Table18.1).
CD123 CAR-T cells induce haematopoietic toxicity but on a smaller scale than
CD33 CAR-T cells, particularly following anti-CD123 single chain fragment vari-
able (scFv) modications (Mardiros et al. 2013; Gill et al. 2014; Thokala et al.
2016). Nevertheless, based on their expression on stem cells, CD123 CAR-T cells
could be used as a myeloablative regimen before ASCT, thus representing an inter-
esting strategy for treatment of R/R AML patients (Gill etal. 2014; Cummins and
Gill 2019; Testa etal. 2019). Notably, IL-15 may enhance the anti-AML activity of
CD123 CAR-T cells (Mu-Mosley etal. 2019). Targeting FLT3 or CD117 could be
an attractive option, again in association with ASCT (Jetani etal. 2018; Myburgh
etal. 2020). Targeting of the Lewis Y antigen and NKG2DL CAR-T cells has also
been proposed, but phase 1 trials have shown short response durations, despite
reduced toxicity (Ritchie etal. 2013; Driouk etal. 2019). CAR-T cells targeting
CD44v6 mediate potent antitumour effects against AML while sparing normal hae-
matopoietic stem cells (Casucci etal. 2013), and a clinical trial is currently ongoing.
A potent effect on LSCs was observed with CAR-T cells targeting IL1RAP (Warda
etal. 2019) with no apparent effect on healthy haematopoietic stem cells. Similar
more specic antileukaemic activity was observed by targeting FLT3 and KIT
mutations (Mitchell etal. 2018). Interestingly, targeting IL1RAP decreases IL-1,
IL-6, IL-10, IL-13, IL-17, IL-22, IFNγ, and TNFα levels (Højen etal. 2019). The
Chemotherapy
Targeted
inhibitors
CAR T-cells
CAR T-cells
Induction
Consolidation
Consolidation
Salvage
Salvage
ASCT
Consolidation
Maintenance
+/- ASCT
HMA
Ta rgeted inhibitor
Immunotherapy
CAR T-cells
HMA
Ta rgeted inhibitor
Immunotherapy
DLI
DLI
HMA
Ta rgeted inhibitor
Immunotherapy
tgTCR T-cells
Salvage ASCT
Maintenance
Maintenance
Relapse
Relapse
CAR T-cells targeting:
• CD33
• CD33/CD123
• CD123
• CD44v6
• CD123/CCL-1
• CD13/TIM-3
• LeY
• NKGD2L
• FLT3
• IL-1RAP
T-cells
CAR
CD33
CD13
TIM-3
CD123
CD44v6
CCL-1
LeY
NKG2DL
IL-1RAP
IL-1RAP
IL-1RAP
IL-1RAP
IL-1RAP
FLT3
KIT
IL-1R
IL-33R
IL-36R
AML blast
AML Cell surface makers
AML tumor burden
AML disease status evolution
Fig. 18.1 Putative place of CAR-T cells in the AML treatment strategy. HMA hypomethylated
agent, DLI donor lymphocyte infusion, tgTCR transgenic T cell receptor T cells, ASCT allogeneic
stem cell transplantation
C. Ferrand and A. Rambaldi

99
reduced production of IL-4, IL-6 and IL-10 and absence of IL-17 production (Warda
etal. 2019) may in turn limit CAR-T cell cytokine release syndrome (CRS) and the
immune effector cell-associated neurotoxicity syndrome (ICAN) associated with
excessive production of IL-1 (Garcia Borrega etal. 2019). Notably, the reduction in
IL-1β, IL-6, and TNFα levels leads to decreased release of IL-10 and TGFβ, which
impair CAR-T cell functions (Epperly etal. 2020).
CAR-T cells simultaneously targeting CD33 and CD123 are also in development
and exhibit pronounced antileukaemic activity (Petrov et al. 2018). Similarly,
CD123 and CCL-1 compound CAR-T cells may be useful for active targeting of
leukaemia stem cells (LSCs) (Morsink etal. 2018; Shang and Zhou 2019).
Table 18.1 CAR-T cell immunotherapies under investigation in AML (based on www.clinicaltri-
als.gov at 05/25/2020)
CAR-T cells
Preclinical results Status
Clinical trials
CD33 Myeloablative, ASCT
requirement
Phase 1 NCT03126864
Phase 1/2 NCT03971799, NCT01864902
CD123 Myeloablative, ASCT
requirement
Phase 1 NCT03796390, NCT03585517,
NCT03114670, NCT03766126,
NCT04014881, NCT03190278,
NCT02159495, NCT04230265,
NCT04318678, NCT03672851
Phase 1/2 NCT04272125, NCT04265963,
NCT04109482, NCT03556982
CCL-1 AML and HSC targeting Phase 1 NCT04219163
CD38 AML targeting Phase 1/2 NCT04351022
CD44v6 AML targeting Phase 1/2 NCT04097301
FLT3 Myeloablative, ASCT
requirement
Phase 1 NCT03904069
KIT (CD117) Myeloablative, ASCT
requirement
Preclinical NCT03473457
B7-H3 HSC toxicity reduction Preclinical None
CD13 TIM-3 HSC toxicity reduction Preclinical None
PD-1 Antitumour enhancement Preclinical None
Lewis Y Short duration of response,
few toxicities
Phase 1 NCT01716364, no further study
NKGD2L Short duration of response,
few toxicities
Phase 1 NCT02203825, no further study
IL1RAP LSC targeting Preclinical NCT04169022
CD33/CD123 AML and HSC targeting Phase 1 NCT04156256
CCL-1/CD123 AML targeting Phase 2/3 NCT03631576
CCL-1/CD33 AML targeting Phase 1 NCT03795779
CCL-1/CD33
and/or CD123
AML targeting Phase 1/2 NCT04010877
Muc1/CLL1/
CD33/CD38/
CD56/ CD123
AML targeting Phase 1/2 NCT03222674
Studies investigating T cell immunotherapies in AML. AML acute myeloid leukaemia, LSC leu-
kaemic stem cell, HSC haematopoietic stem cell, ASCT allogeneic stem cell transplantation
18 Myeloid Malignancies

100
Bispecic CD13-TIM-3 CAR-T cells (He etal. 2020) and B7-H3 CAR-T cells
(Lichtman etal. 2018) showed reduced HSC toxicity. Moreover, the B7-H3 pan-
cancer target was also studied in solid tumours (Waldman etal. 2020). Preliminary
reports show that PD-1 inhibitors also regulate the CAR-T cell response, although
few data are available. Furthermore, delivery of PD-1-blocking scFv CAR-T cells
in preclinical investigations demonstrated interesting antitumour efcacy enhance-
ment (Anonymous 2019). Several challenges remain to be overcome, as recently
reported, and further investigations may provide a better understanding (Mardiana
and Gill 2020).
Molecular Engineering oftheChimeric Receptor
andAlternative Cell Sources
Beyond the selected target, optimizing the molecular engineering of the chimeric
receptor remains crucial. CD33 4-1BBz CAR-T cells have shown antileukaemic
activity and resistance to exhaustion with increasing central memory comportment
(Li etal. 2018). An additional strategy that has been proposed to reduce haemato-
poietic toxicity is the use of a transiently expressed CART33 to induce self-limiting
activity against AML cells (Kenderian etal. 2015). Another proposed strategy is to
inactivate the CD33 gene in HSCs prior to transplantation to prevent CD33-induced
haematopoietic toxicity of CAR-T cells (Kim etal. 2018).
In addition, to avoid or reduce the uncontrolled toxicity of expanding CAR-T
cells, the use of the anti-CD52 antibody alemtuzumab or a suicide gene strategy
based on CD20 protein coexpression in CD123 CAR-T cells has been proposed for
subsequent anti-CD20 targeting with rituximab (Introna et al. 2000; Tasian
etal. 2017).
Several clinical trials are currently evaluating the use of allogeneic CAR-T cells
in haematologic malignancies, employing different effector cell types, such as NK
cells (Daher and Rezvani 2021) or TCR-edited cells (Provasi etal. 2012), to limit
GvHD and develop strategies to avoid the rejection of allogeneic cells. In this
regard, the limited GvHD associated with the use of cytokine-induced killer (CIK)
cells (Martino Introna etal. 2017) was conrmed in a phase I/IIa study in which
B-ALL patients who relapsed after allogeneic transplantation were treated using
CD19-specic CAR CIK cells (CARCIK-CD19) manufactured from a previous
transplant donor (Magnani etal. 2020). Notably, this study provides evidence of the
feasibility of employing a nonviral sleeping beauty transposon system to success-
fully produce CARCIK cell products starting from a small amount of donor-derived
PB, thus offering a valid alternative to viral vectors. The use of CAR-engineered
CIK cells was also demonstrated to be effective for AML by characterizing the tar-
geting of the two most validated AML molecules, CD33 and CD123, invitro and
invivo (Tettamanti etal. 2013; Pizzitola etal. 2014; Arcangeli etal. 2017; Rotiroti
etal. 2020).
C. Ferrand and A. Rambaldi

101
References
Anonymous. Augmenting CAR-T cells with PD-1 blockade. Cancer Discov. 2019;9(2): 158.
https://doi.org/10.1158/2159- 8290.CD- NB2018- 165.
Arcangeli S, Rotiroti MC, Bardelli M, Simonelli L, Magnani CF, Biondi A, Biagi E, Tettamanti S,
Varani L.Balance of anti-CD123 chimeric antigen receptor binding afnity and density for the
targeting of acute myeloid leukemia. Mol Ther. 2017;25(8):1933–45. https://doi.org/10.1016/j.
ymthe.2017.04.017.
Borrega G, Jorge PG, Rüger MA, Onur ÖA, Shimabukuro-Vornhagen A, Kochanek M, Böll
B.In the eye of the storm: immune-mediated toxicities associated with CAR-T cell therapy.
HemaSphere. 2019;3(2):e191. https://doi.org/10.1097/HS9.0000000000000191.
Casucci M, di Robilant BN, Falcone L, Camisa B, Norelli M, Genovese P, Gentner B, etal. CD44v6-
targeted T cells mediate potent antitumor effects against acute myeloid leukemia and multiple
myeloma. Blood. 2013;122(20):3461–72. https://doi.org/10.1182/blood- 2013- 04- 493361.
Cummins KD, Gill S. Chimeric antigen receptor T-cell therapy for acute myeloid leuke-
mia: how close to reality? Haematologica. 2019;104(7):1302–8. https://doi.org/10.3324/
haematol.2018.208751.
Daher M, Rezvani K.Outlook for new CAR-based therapies with a focus on CAR NK cells:
what lies beyond CAR-engineered T cells in the race against cancer. Cancer Discov.
2021;11(1):45–58. https://doi.org/10.1158/2159- 8290.CD- 20- 0556.
Driouk L, Gicobi J, Kamihara Y, Rutherford K, Dranoff G, Ritz J, Baumeister SHC.Chimeric
antigen receptor T cells targeting NKG2D-ligands show robust efcacy against acute myeloid
leukemia and T-cell acute lymphoblastic leukemia. Blood. 2019;134(Suppl 1):1930. https://
doi.org/10.1182/blood- 2019- 130113.
Epperly R, Gottschalk S, Paulina Velasquez M.A bump in the road: how the hostile AML micro-
environment affects CAR-T cell therapy. Front Oncol. 2020;10:262. https://doi.org/10.3389/
fonc.2020.00262.
Gill S, Tasian SK, Ruella M, Shestova O, Li Y, Porter DL, Carroll M, etal. Preclinical targeting of
human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modied T
cells. Blood. 2014;123(15):2343–54. https://doi.org/10.1182/blood- 2013- 09- 529537.
He X, Feng Z, Ma J, Ling S, Cao Y, Gurung B, Wu Y, etal. Bispecic and split CAR-T cells target-
ing CD13 and TIM3 eradicate acute myeloid leukemia. Blood. 2020;135(10):713–23. https://
doi.org/10.1182/blood.2019002779.
Hofmann S, Schubert M-L, Wang L, He B, Neuber B, Dreger P, Müller-Tidow C, Schmitt
M.Chimeric antigen receptor (CAR) T cell therapy in acute myeloid leukemia (AML). J Clin
Med. 2019;8(2):200. https://doi.org/10.3390/jcm8020200.
Højen JF, Kristensen MLV, McKee AS, Wade MT, Azam T, Lunding LP, de Graaf DM, et al.
IL-1R3 blockade broadly attenuates the functions of six members of the IL-1 family, reveal-
Key Points
• The primary challenge limiting the use of CAR-T cells in myeloid malig-
nancies is the absence of an ideal antigen.
• Myeloid antigens are often coexpressed on normal haematopoietic stem/
progenitor cells (HSPCs).
• Myelotoxicity and endothelial toxicity can be overcome by “and/or” dual
CAR targeting.
• Allogeneic CAR-T cells may be a future alternative.
18 Myeloid Malignancies

102
ing their contribution to models of disease. Nat Immunol. 2019;20(9):1138–49. https://doi.
org/10.1038/s41590- 019- 0467- 1.
Introna M, Barbui AM, Bambacioni F, Casati C, Gaipa G, Borleri G, Bernasconi S, et al.
Genetic modication of human T cells with CD20: a strategy to purify and lyse trans-
duced cells with anti-CD20 antibodies. Hum Gene Ther. 2000;11(4):611–20. https://doi.
org/10.1089/10430340050015798.
Introna M, Lussana F, Algarotti A, Gotti E, Valgardsdottir R, Mico C, Grassi A, etal. Phase II
study of sequential infusion of donor lymphocyte infusion and cytokine-induced killer cells for
patients relapsed after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow
Transplant. 2017;23(12):2070–8. https://doi.org/10.1016/j.bbmt.2017.07.005.
Jetani H, Garcia-Cadenas I, Nerreter T, Thomas S, Rydzek J, Meijide JB, Bonig H, etal. CAR-T
cells targeting FLT3 have potent activity against FLT3(−)ITD(+) AML and act synergistically
with the FLT3-inhibitor crenolanib. Leukemia. 2018;32(5):1168–79. https://doi.org/10.1038/
s41375- 018- 0009- 0.
Kenderian SS, Ruella M, Shestova O, Klichinsky M, Aikawa V, Morrissette JJD, Scholler J, etal.
CD33-specic chimeric antigen receptor T cells exhibit potent preclinical activity against
human acute myeloid leukemia. Leukemia. 2015;29(8):1637–47. https://doi.org/10.1038/
leu.2015.52.
Kim MY, Kyung-Rok Y, Kenderian SS, Ruella M, Chen S, Shin T-H, Aljanahi AA, etal. Genetic
inactivation of CD33 in hematopoietic stem cells to enable CAR-T cell immunotherapy
for acute myeloid leukemia. Cell. 2018;173(6):1439–1453.e19. https://doi.org/10.1016/j.
cell.2018.05.013.
Li S, Tao Z, Yingxi X, Liu J, An N, Wang Y, Xing H, etal. CD33-specic chimeric antigen receptor
T cells with different co-stimulators showed potent anti-leukemia efcacy and different pheno-
type. Hum Gene Ther. 2018;29(5):626–39. https://doi.org/10.1089/hum.2017.241.
Lichtman E, Hongwei D, Savoldo B, Ferrone S, Li G, Lishan S, Dotti G.Pre-clinical evaluation of
B7-H3-specic chimeric antigen receptor T-cells for the treatment of acute myeloid leukemia.
Blood. 2018;132(Suppl 1):701. https://doi.org/10.1182/blood- 2018- 99- 113468.
Magnani CF, Gaipa G, Lussana F, Belotti D, Gritti G, Napolitano S, Matera G, etal. Sleeping
beauty–engineered CAR-T cells achieve antileukemic activity without severe toxicities. J Clin
Invest. 2020;130(11):6021–33. https://doi.org/10.1172/JCI138473.
Mardiana S, Gill S.CAR-T cells for acute myeloid leukemia: state of the art and future directions.
Front Oncol. 2020;10:697. https://doi.org/10.3389/fonc.2020.00697.
Mardiros A, Dos Santos C, McDonald T, Brown CE, Xiuli Wang L, Budde E, Hoffman L,
et al. T cells expressing CD123-specic chimeric antigen receptors exhibit specic cyto-
lytic effector functions and antitumor effects against human acute myeloid leukemia. Blood.
2013;122(18):3138–48. https://doi.org/10.1182/blood- 2012- 12- 474056.
Mitchell K, Barreyro L, Todorova TI, Taylor SJ, Antony-Debré I, Narayanagari S-R, Carvajal
LA, etal. IL1RAP potentiates multiple oncogenic signaling pathways in AML.J Exp Med.
2018;215(6):1709–27. https://doi.org/10.1084/jem.20180147.
Morsink L, Walter R, Ossenkoppele G.Prognostic and therapeutic role of CLEC12A in acute
myeloid leukemia. Blood Rev. 2018;34:26–33. https://doi.org/10.1016/j.blre.2018.10.003.
Mu-Mosley H, Ostermann LB, Muftuoglu M, Schober W, Patel NB, Vaidya A, Bonifant CL,
Gottschalk S, Velasquez MP, Andreeff M.Transgenic expression of IL15in CD123-specic
BiTE-secreting engager T-cells results in improved anti-AML activity. Blood. 2019;134(Suppl
1):3917. https://doi.org/10.1182/blood- 2019- 125928.
Myburgh R, Kiefer JD, Russkamp NF, Magnani CF, Nuñez N, Simonis A, Pster S, etal. Anti-human
CD117 CAR-T cells efciently eliminate healthy and malignant CD117-expressing hemato-
poietic cells. Leukemia. 2020;34(10):2688–703. https://doi.org/10.1038/s41375- 020- 0818- 9.
Petrov JC, Wada M, Pinz KG, Yan LE, Chen KH, Shuai X, Liu H, etal. Compound CAR-T cells as a
double-pronged approach for treating acute myeloid leukemia. Leukemia. 2018;32(6):1317–26.
https://doi.org/10.1038/s41375- 018- 0075- 3.
Pizzitola I, Anjos-Afonso F, Rouault-Pierre K, Lassailly F, Tettamanti S, Spinelli O, Biondi A,
Biagi E, Bonnet D.Chimeric antigen receptors against CD33/CD123 antigens efciently target
C. Ferrand and A. Rambaldi

103
primary acute myeloid leukemia cells invivo. Leukemia. 2014;28(8):1596–605. https://doi.
org/10.1038/leu.2014.62.
Provasi E, Genovese P, Lombardo A, Magnani Z, Liu P-Q, Reik A, Chu V, etal. Editing T cell
specicity towards leukemia by zinc nger nucleases and lentiviral gene transfer. Nat Med.
2012;18(5):807–15. https://doi.org/10.1038/nm.2700.
Ritchie DS, Neeson PJ, Khot A, Peinert S, Tai T, Tainton K, Chen K, etal. Persistence and efcacy
of second generation CAR-T cell against the LeY antigen in acute myeloid leukemia. Mol
Ther. 2013;21(11):2122–9. https://doi.org/10.1038/mt.2013.154.
Rotiroti MC, Buracchi C, Arcangeli S, Galimberti S, Valsecchi MG, Perriello VM, Rasko T,
etal. Targeting CD33in chemoresistant AML patient-derived xenografts by CAR-CIK cells
modied with an improved SB transposon system. Mol Ther. 2020;28(9):1974–86. https://doi.
org/10.1016/j.ymthe.2020.05.021.
Roussel X, Daguindau E, Berceanu A, Desbrosses Y, Warda W, Neto M, da Rocha R, Trad
ED, Deschamps M, Ferrand C.Acute myeloid leukemia: from biology to clinical practices
through development and pre-clinical therapeutics. Front Oncol. 2020;10:599933. https://doi.
org/10.3389/fonc.2020.599933.
Shang Y, Zhou F.Current advances in immunotherapy for acute leukemia: an overview of antibody,
chimeric antigen receptor, immune checkpoint, and natural killer. Front Oncol. 2019;9:917. https://
doi.org/10.3389/fonc.2019.00917.
Tasian SK, Kenderian SS, Shen F, Ruella M, Shestova O, Kozlowski M, Li Y, etal. Optimized deple-
tion of chimeric antigen receptor T cells in murine xenograft models of human acute myeloid
leukemia. Blood. 2017;129(17):2395–407. https://doi.org/10.1182/blood- 2016- 08- 736041.
Testa U, Pelosi E, Castelli G.CD123 as a therapeutic target in the treatment of hematological
malignancies. Cancers. 2019;11(9):1358. https://doi.org/10.3390/cancers11091358.
Tettamanti S, Marin V, Pizzitola I, Magnani CF, Giordano GMP, Attianese EC, Maltese F, etal.
Targeting of acute myeloid leukaemia by cytokine-induced killer cells redirected with a novel
CD123-specic chimeric antigen receptor. Br J Haematol. 2013;161(3):389–401. https://doi.
org/10.1111/bjh.12282.
Thokala R, Olivares S, Mi T, Maiti S, Deniger D, Huls H, Torikai H, etal. Redirecting specicity
of T cells using the sleeping beauty system to express chimeric antigen receptors by mix-and-
matching of VL and VH domains targeting CD123+ tumors. PLoS One. 2016;11(8):e0159477.
Waldman AD, Fritz JM, Lenardo MJ.A guide to cancer immunotherapy: from T cell basic sci-
ence to clinical practice. Nat Rev Immunol. 2020;20(11):651–68. https://doi.org/10.1038/
s41577- 020- 0306- 5.
Warda W, Larosa F, Da Rocha MN, Trad R, Deconinck E, Fajloun Z, Faure C, etal. CML hemato-
poietic stem cells expressing IL1RAP can be targeted by chimeric antigen receptor–engineered
T cells. Cancer Res. 2019;79(3):663–75. https://doi.org/10.1158/0008- 5472.CAN- 18- 1078.
Open Access
This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
18 Myeloid Malignancies

105
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_19
P. Pedrazzoli
Oncology Unit, Fondazione IRCCS Policlinico San Matteo, Department of Internal Medicine
and Medical Therapy, University of Pavia, Pavia, Italy
e-mail: [email protected].it
J. B. A. G. Haanen (
*)
Divisions of Medical Oncology and Molecular Oncology & Immunology, The Netherlands
Cancer Institute, Amsterdam, The Netherlands
e-mail: [email protected]
19
Developments inSolid Tumours
PaoloPedrazzoli andJohnB.A.G.Haanen
Chimeric antigen receptor (CAR) T cells have emerged as breakthrough therapies in
patients with refractory haematologic malignancies, and the highly encouraging
clinical results have fuelled expectations of implementing these strategies in other
cancer types. However, a similar success of CAR-T cell treatment has not yet been
observed in solid tumours. Various factors, including the immunosuppressive nature
of the tumour microenvironment, hinder of CAR-T cell trafcking and inltration
into scarcely accessible tumour sites, and difculties in identifying targetable anti-
gens with optimal expression and a good toxicity prole, and limiting CAR-T dose
escalation, must be overcome to achieve success in the treatment of solid cancers
(Comoli etal. 2019).
Several clinical trials have tested the efcacy of CAR-T cells in solid tumours.
To date, clinical results have not been encouraging, with a general lack of therapeu-
tic response and the presence of on-target off-tumour toxicity. However, some stud-
ies have achieved promising outcomes that justify further exploration of this
approach in solid tumours, as is happening in many areas of the world (Comoli
etal. 2019).
Early experiences with GD2-specic CAR-T cells showed objective responses in
paediatric patients with neuroblastoma (Pule etal. 2008). Since then, lymphocytes
have been engineered via insertion of third-generation CARs and, more recently, by
delivery of a GD2-CAR-IL-15 construct to NK cells (Heczey etal. 2020).

106
Similarly, the safety and antitumour activity of CAR-T cells targeting a variety
of antigens, such as IL-13Rα2, epidermal growth factor receptor-vIII, and human
epidermal growth factor receptor-2 (HER2), have been assessed in patients with
glioblastoma multiforme (GBM). Infusion of second-generation CD28ζ HER2-
specic CAR-modied virus-specic T cells was well tolerated, with no dose-
limiting toxic effects, and led to clinical benet; 1 patient showed a partial response
(PR) lasting more than 9months, and 7 patients had stable disease (SD) for several
months (Ahmed etal. 2017). Other clinical trials have demonstrated the feasibility,
safety, and clinical efcacy of second-generation EGFRvIII-specic and IL13BBζ–
specic CAR-T cells (Brown etal. 2016) in patients with refractory GBM.
In gastrointestinal neoplasms, a clinical trial utilizing CEA CAR-T therapy in ten
patients with metastatic colorectal cancer (CRC) resulted in SD in seven patients,
without severe adverse events related to CAR-T therapy (Zhang etal. 2017). A pre-
vious case report of HER2-specic cell therapy for CRC using third generation
CAR-T cells caused fatal acute respiratory distress syndrome due to recognition of
lung epithelial cells expressing low levels of HER2 (Morgan etal. 2010). A phase I
study of second-generation CAR-T cells targeting HER2 was conducted in 11
patients with advanced biliary tract cancer or pancreatic cancer. A 4.5-month partial
response was observed, and 5 subjects achieved stable disease. Toxicity was man-
ageable, with grade 3 fever and one patient showing elevation of liver enzymes as
CAR-T-related adverse events; one episode of reversible severe upper gastrointesti-
nal haemorrhage occurred in a patient with gastric involvement 11days after the
HER2 CAR-T- cell infusion, and 2 cases of grade 1–2 delayed fever accompanied
by increases in C-reactive protein and interleukin-6 were observed (Feng et al.
2018). Epidermal growth factor receptor (EGFR) and CD133-specic CAR-T
sequential immunotherapy were employed by the same group in a patient with
advanced unresectable/metastatic cholangiocarcinoma (CCA), resulting in a PR
lasting more than 12 months; however, slight liver toxicity secondary to EGFR
CAR-T therapy and epidermal and endothelial damage due to CD133-specic
CAR-T immunotherapy was observed.
Similar to these experiences, second-generation HER2-specic CAR-T cells,
used in a phase I clinical trial conducted on 19 patients with refractory HER2-
positive sarcoma, induced SD lasting from 12weeks to 14months in 4 of the evalu-
able patients (Ahmed etal. 2015).
Investigators at the University of Pennsylvania explored an approach based on
mRNA-transduced CAR-T cells that target mesothelin (meso CAR-T) in patients
with advanced malignant pleural mesothelioma (MPM) or advanced pancreatic can-
cer. In the rst two patients reported, meso CAR-T cells showed some antitumour
activity in vivo in the absence of distinct toxicities (Beatty et al. 2014). Second-
generation CAR-T cells specic for EGFR were employed in a phase I study to treat
11 patients with advanced non-small-cell lung cancer (NSCLC), resulting in 2 PR
cases and 5 SD cases, lasting from 2 to 8months, with limited adverse events, includ-
ing skin toxicity, nausea, vomiting, dyspnoea, and hypotension (Feng etal. 2016).
A few phase I studies and case series have reported CAR-T cell treatment for
other solid tumours, such as melanoma, breast cancer, renal cell carcinoma, prostate
cancer, and ovarian and seminal vesicle cancer (reviewed Fucà etal. 2020).
P. Pedrazzoli and J. B. A. G. Haanen

107
Various toxicities were observed after CAR-T cell infusion for treatment of solid
tumours. In the setting of haematologic malignancies, cytokine release syndrome
(CRS) is a frequent, potentially severe adverse event following CAR-T cell therapy.
However, CRS, as well as the neurological toxicity (ICAN) sometimes observed in
the haematologic setting, has not yet become a common event after CAR-T cell ther-
apy for solid tumours, perhaps because of the lower tumour load. Conversely, CAR-T
cells trials conducted in solid tumour cohorts showed critical, unexpected on-target,
off-tumour toxicities resulting from the recognition by CAR-T cells of tumour anti-
gens expressed on healthy tissues (Morgan etal. 2010; Lamers etal. 2013). Targeting
tumour-specic antigens appears to result in fewer off-tumour effects, but whether
these CAR-T cells have promising clinical efcacy remains to be seen, and many tri-
als are still ongoing. Strategies to increase tumour selectivity while sparing healthy
tissues are being evaluated to control on-target off-tumour toxicity.
Despite novel genetic engineering techniques and combinatorial approaches to
counteract biological barriers, tumour heterogeneity, and the immunosuppressive
properties of the tumour microenvironment, targeting solid tumours with CAR-T
cells in the clinical setting remains challenging. Therefore, T cell therapy, alone or
in combination with immune checkpoint inhibitors or other agents targeting either
the cancer cell or the tumour environment, will likely play a role in improving can-
cer treatment outcomes (Apetoh et al. 2015). Designing and selecting the most
appropriate clinical trials or settings to rapidly identify combinatorial approaches
that are efcacious in different patient populations and identifying patients who will
best benet from immune checkpoint inhibitors alone (Chalabi etal. 2020) or from
the addition of other targeted immunotherapies will be the most pressing need for
the future success of CAR immunotherapy in solid cancer.
References
Ahmed N, Brawley VS, Hegde M, et al. Human epidermal growth factor receptor 2 (HER2)-
specic chimeric antigen receptor-modied T cells for the immunotherapy of HER2-positive
sarcoma. J Clin Oncol. 2015;20(33):1688–96.
Ahmed N, Brawley V, Hegde M, etal. HER2-specic chimeric antigen receptor-modied virus-
specic T cells for progressive glioblastoma: a phase 1 dose-escalation trial. JAMA Oncol.
2017;3:1094–101.
Key Points
• CAR-T therapy is under development for many solid cancer types, but
important breakthroughs have not yet been achieved.
• Tumour heterogeneity, the immunosuppressive tumour microenvironment,
and other barriers are hurdles that must be overcome before CAR-T cells
can be effective against solid cancers.
• The eld may benet from network models for CAR-T cell production in
academic centres.
19 Developments inSolid Tumours

108
Apetoh L, Ladoire S, Coukos G, etal. Combining immunotherapy and anticancer agents: the right
path to achieve cancer cure? Ann Oncol. 2015;26:1813–23.
Beatty GL, Haas AR, Maus MV, et al. Mesothelin-specic chimeric antigen receptor mRNA-
engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res.
2014;2:112–20.
Brown C, Alizadeh D, Starr R, etal. Regression of glioblastoma after chimeric antigen receptor
T-cell therapy. N Engl J Med. 2016;375:2561–9.
Chalabi M, Fanchi LF, Dijkstra KK, et al. Neoadjuvant immunotherapy leads to pathologi-
cal responses in MMR-procient and MMR-decient early-stage colon cancers. Nat Med.
2020;26:566–76.
Comoli P, Chabannon C, Koehl U, Lanza F, Urbano-Ispizua A, Hudecek M, Ruggeri A, Secondino
S, Bonini C, Pedrazzoli P.Development of adaptive immune effector therapies in solid tumors.
Ann Oncol. 2019;30:1740–50.
Feng K, Guo Y, Dai H, etal. Chimeric antigen receptor-modied T cells for the immunotherapy
of patients with EGFR expressing advanced relapsed/refractory non-small cell lung cancer. Sci
China Life Sci. 2016;59:468–79.
Feng K, Liu Y, Guo Y, Qiu J, Wu Z, Dai H, etal. Phase I study of chimeric antigen receptor modi-
ed T cells in treating HER2-positive advanced biliary tract cancers and pancreatic cancers.
Protein Cell. 2018;9:838–47.
Fucà G, Reppel L, Landoni E, Savoldo B, Dotti G.Enhancing chimeric antigen receptor T-cell
efcacy in solid tumors. Clin Cancer Res. 2020;26:2444–51.
Heczey A, Courtney AN, Montalbano A, Robinson S, Liu K, Li M, Ghatwai N, Dakhova O, Liu
B, Raveh-Sadka T, Chauvin-Fleurence CN, Xu X, Ngai H, Di Pierro EJ, Savoldo B, Dotti G,
Metelitsa LS. Anti-GD2 CAR-NKT cells in patients with relapsed or refractory neuroblas-
toma: an interim analysis. Nat Med. 2020;26:1686–90.
Lamers CH, Sleijfer S, van Steenbergen S, etal. Treatment of metastatic renal cell carcinoma with
CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol
Ther. 2013;21:904–12.
Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA.Case report of a seri-
ous adverse event following the administration of T cells transduced with a chimeric antigen
receptor recognizing ERBB2. Mol Ther. 2010;18:843–51.
Pule M, Savoldo B, Myers GD, etal. Virus-specic T cells engineered to coexpress tumor-specic
receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med.
2008;14:1264–70.
Zhang C, Wang Z, Yang Z, etal. Phase I escalating-dose trial of CAR-T therapy targeting CEA(+)
metastatic colorectal cancers. Mol Ther. 2017;25:1248–58.
Open Access
This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
P. Pedrazzoli and J. B. A. G. Haanen

Part IV
Clinical Management of Patients
Treated with CAR-T Cells

111
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_20
N. Boissel
URP-3518, Institut de Recherche Saint-Louis, Université de Paris, Paris, France
Service d’Hématologie Adolescents et Jeunes Adultes, Assistance Publique des Hôpitaux de
Paris, Paris, France
e-mail: [email protected]
F. Ciceri (
*)
Vita-Salute San Raffaele University, Milan, Italy
Hematology and Bone Marrow Transplantation Unit, IRCCS San Raffaele Hospital,
Milan, Italy
e-mail: ciceri.fabio@hsr.it
20
Bridging Chemotherapy: Adult Acute
Lymphoblastic Leukaemia
NicolasBoissel andFabioCiceri
Bridging therapy can be given after leukapheresis and before lymphodepletion dur-
ing CAR-T cell manufacturing. The primary goal of bridging therapies is to prevent
uncontrolled progression of the underlying disease during the manufacturing period
before CAR-T cell infusion. Several studies indicate that a high tumour burden is
associated with an increased risk of complications after CAR-T cell infusion (Cohen
et al. 2019). Therefore, controlling the disease and even possibly decreasing the
tumour burden is critical during the manufacturing period. The choice of bridging
therapies is essential for the success of the procedure.
Clinical trials of CD19 CAR-T therapy in B-ALL reproducibly report high rates
of patient dropout after enrolment due to disease progression or treatment-related
complications (Park etal. 2018; Maude etal. 2018). For example, among 75 patients
who received a CAR-T infusion in the ELIANA study, 65 (87%) were treated with
bridging chemotherapy between enrolment and infusion, and 10 out of 92 patients
enrolled in the trial could not be infused due to signicant adverse events or death
(Maude et al. 2018). The rate of adult patients infused in the Memorial Sloan
Kettering (MSKCC) experience was 65% (54/83, 65%) of enrolled patients, mostly
due to disease progression and death (Park etal. 2018). This reects the challenges
in clinical management during the 3–6-week period necessary for autologous
CAR-T cell preparation (the bridging period).

112
Given that CD19 CAR-T therapies are currently indicated for relapsed/refractory
B-ALL patients who have already been exposed to one or more lines of potentially
effective therapies, often including combinations of several agents, and that these
patients often require therapeutic intervention against rapidly progressive disease or
a high tumour burden, the choice of the better approach is not trivial.
Several bridging therapy options now exist, including high-intensity chemo-
therapy, targeted agents (e.g., TKIs), immunotherapies (e.g., CD-19 or CD22-
directed), and low-intensity approaches (e.g., vincristine, 6-MP, steroids,
thioguanine, etc.). Each approach has pros and cons. For example, high-intensity
chemotherapy might be too toxic to allow treatment with CAR-T cells to pro-
ceed, while low-intensity approaches might fail in terms of tumour burden
reduction.
In addition, treatment with CD19-directed therapies, such as the bispecic T cell
engager blinatumomab, might have an impact on the efcacy of subsequent CD19
CAR-T cell therapy (Pillai etal. 2019), and common mechanisms of tumour escape
to CD19-directed therapies have now been reported (Boissel 2021). Blinatumomab
use was an exclusion criterion from the ELIANA trial (Maude etal. 2018), while it
was allowed for patients participating in other similar trials. In the expanded access
program for tisagenlecleucel, the overall response rate in patients with prior blina-
tumomab treatment was 67% versus 90% in other patients (Baruchel etal. 2020).
However, no univocal data on this important salvage option are available in this
setting.
In a recent study, the Memorial Sloan Kettering group reviewed different
bridging strategies and outcomes for all patients enrolled in a single-centre,
phase 1 trial of CD19-specific CAR-T cells for R/R adult ALL (ClinicalTrials.
gov NCT01044069) (Perica etal. 2021). They observed that reductions in dis-
ease burden during the bridging period are associated with favourable out-
comes after CAR-T therapy and thus suggest that optimal strategies to reduce
disease burden during bridging are warranted. They proposed a bridging strat-
egy based on disease burden at the time of the CAR-T therapy decision. They
recommended low-intensity therapy for patients with a low tumour burden,
low-intensity chemotherapy, or targeted therapy (e.g., inotuzumab) for patients
with a high disease burden who are chemorefractory (e.g., partial or short
response to prior line of chemotherapy) and unlikely to benefit from high-
intensity bridging, and a careful evaluation of the risks and benefits of high- vs.
low-intensity therapy for patients with high disease burden with expected che-
mosensitivity (e.g., limited prior chemotherapy exposure, late relapse, or sen-
sitivity to the last line). In fact, not surprisingly, the study showed an increased
rate of infections during the bridging period in the high-intensity chemother-
apy group.
In conclusion, tumour burden, patient comorbidities, and disease characteristics
should tailor the choice of the optimal bridging therapy. The goal of this therapy is
not complete disease eradication per se but reduction of tumour burden, preserving
N. Boissel and F. Ciceri

113
the patient in good clinical condition for CAR-T cell infusion. The benets of
tumour burden reduction may be twofold, with (1) a reduction in the early risk of
adverse events, including cytokine release syndrome and (2) a better outcome after
CAR-T cell therapy. Although the role of B-cell-directed therapies should be further
and carefully investigated in this setting, mainly to exclude possible interference
with CAR-T cell expansion or activity, targeted and low-intensity approaches could
be instrumental for this objective. Conversely, high-intensity chemotherapy should
be limited to those cases in which the benet and the probability of achieving a
rapid tumour load reduction overcome the risk of infection or another toxic event in
the context of a CAR-T-oriented strategy.
References
Baruchel A, Krueger J, Balduzzi A, etal. Tisagenlecleucel for pediatric/young adults with relapsed/
refractory acute lymphoblastic leukemia: preliminary report on B2001X study focusing on
prior exposure to blinatumomab and inotuzumab. J Clin Oncol. 2020;38(15 Suppl):10518.
Boissel N.ALL in escape room. Blood. 2021;137(4):432–4.
Cohen AD, Garfall AL, Stadtmauer EA, etal. B cell maturation antigen-specic CAR-T cells are
clinically active in multiple myeloma. J Clin Invest. 2019;129(6):2210–21.
Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, etal. Tisagenlecleucel in
children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378:439–48.
Park JH, Rivière I, Gonen M, etal. Long-term follow-up of CD19 CAR-therapy in acute lympho-
blastic leukemia. N Engl J Med. 2018;378:449–59.
Perica K, Flynn J, Curran KJ, etal. Impact of bridging chemotherapy on clinical outcome of CD19
CAR-T therapy in adult acute lymphoblastic leukemia. Leukemia. 2021;35(11):3268–71.
Pillai V, Muralidharan K, Meng W, et al. CAR-T cell therapy is effective for CD19-dim
B-lymphoblastic leukemia but is impacted by prior blinatumomab therapy. Blood Adv.
2019;3:3539–49.
Further Reading
Anagnostou T, Riaz IB, Hashmiet SK, etal. Anti-CD19 chimeric antigen receptor T-cell therapy
in acute lymphocytic leukaemia: a systematic review and metaanalysis. Lancet Haematol.
2020;7:e816–26.
Brentjens RJ, Rivière I, Park JH, etal. Safety and persistence of adoptively transferred autologous
CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias.
Blood. 2011;118:4817–28.
Davila ML, Riviere I, Wang X, etal. Efcacy and toxicity management of 19-28z CAR-T cell
therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6:224ra25.
Key Point
• Disease control is necessary before CAR-T cell infusion.
20 Bridging Chemotherapy: Adult Acute Lymphoblastic Leukaemia

114
Lee DW, Kochenderfer JN, Stetler-Stevenson M, etal. T cells expressing CD19 chimeric antigen
receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-
escalation trial. Lancet. 2015;385:517–28.
Maude SL, Frey N, Shaw PA, etal. Chimeric antigen receptor T cells for sustained remissions in
leukemia. N Engl J Med. 2014;371:1507–17.
Open Access
This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
N. Boissel and F. Ciceri

115
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_21
A. Baruchel (*)
Université de Paris et EA 3518 Institut de Recherche Saint-Louis, Paris, France
Service d’Hémato-Immunologie Pédiatrique, Hôpital Universitaire Robert Debré (APHP),
Paris, France
e-mail: andre.baruchel@rdb.aphp.fr
21
Bridging toCAR-T Cells inChildren,
Adolescents, andYoung Adults withALL
AndréBaruchel
The Importance oftheBridge
Leukapheresis can be performed in most patients, including infants, but not all
patients will receive autologous CAR-T cells. The ELIANA trial can be taken as an
example to help understand the issues: 97 patients were successfully screened and
enrolled, but only 79 of them nally made it to the infusion. Of the remaining 18
patients, 10 died or experienced an AE during the manufacturing time, and 8 patients
had issues with the manufacturing process (Grupp etal. 2018).
Contrary to what is expected prior to allogeneic haematopoietic cell transplanta-
tion (allo-HCT), the role of an optimal bridging therapy is not to obtain the lowest
residual disease but only a reduction or stabilization of tumour burden, bringing the
patient to the CAR-T cell infusion in good clinical condition.
Some facts are to recall:
• The interval between apheresis and infusion is highly variable and during clini-
cal trials can range between 3weeks and 3months. With the approved commer-
cial product tisagenlecleucel, the interval is now in the 3–4-week range. Of note,
manufacturing could be shorter in academic closed systems using fresh cells and
decentralized manufacturing.
• Many variables can indeed inuence the nal interval:
– Cryopreservation to shipping: time can be lost, being inuenced by manufac-
turing slots.
– Manufacturing site: USA vs. EU.

116
– Availability of the cell therapy lab to receive the apheresis product and then the
common availability of the same lab and the pharmacists to receive the “drug”.
– Availability of a room in the clinical department and the possibility that the
patient can be admitted to the ICU in the case of complications, which can be
an issue in the current pandemic.
– Of utmost importance are the course of the disease and the clinical status,
with the main risks being disease progression, occurrence of infection (fungal
diseases in particular) in these heavily pretreated patients and other SAEs
linked to chemo/immunotherapies (e.g., inotuzumab).
Thus, the « art of bridging » includes the following steps:
• Selecting the best chemotherapy requires integrating the disease biology, previ-
ous disease sensitivity to treatment and the tolerance history of the patient. There
is no « one size ts all » here.
• Aiming to undergo lymphodepletion plus infusion without too much disease as a
way to decrease the incidence of CRS and increase the nal outcome.
• Monitoring a patient who is not completely under your control: close collaboration
and numerous contacts (at least 2/week) with the referring centre are mandatory.
Examples of possible choices are found in Table21.1.
Table 21.1 Possible bridging therapies on the road to CAR-T cell therapy according to tumour
burden and disease localization and kinetics
a
No treatment: smouldering disease
Low-intensity chemotherapy: low disease burden and/or slowly progressing ALL
• Weekly vincristine (VCR) with oral 6MP and methotrexate (MTX).
• Weekly VCR plus dexamethasone (DEX) 6mg/m
2
2 days/week.
Intermediate-intensity chemotherapy: disease burden and/or progressing ALL
• Consolidation « IB » (6MP, cytarabine, cyclophosphamide).
• Weekly VCR plus DEX, bortezomib, asparaginase.
High-intensity chemotherapy: aggressive disease or EMD
b
• High-dose (HD) cytarabine, VP16-cyclophosphamide, hyper CVAD.
• High-dose MTX if CNS involvement.
Very high-intensity chemotherapy for rapidly progressing disease:
• Sequential approach, e.g., HD cytarabine followed by LD.
a
Targeted agents, such as TKIs, can be used in Ph+ ALL and ABL class fusion ALL in addition to
low-intensity chemotherapy, for example
b
EMD: extramedullary disease
A. Baruchel

117
Is There aPlace forImmunotherapy?
• Accumulating data suggest that the use of CD19-oriented therapy (e.g., blinatu-
momab) prior to administration of CD19 CAR-T cells can be detrimental, par-
ticularly with the selection of CD19-negative clones (Pillai etal. 2019; Baruchel
etal. 2020; Taraseviciute etal. 2020).
• Inotuzumab ozogamicin, an anti-CD22 therapy, can be used in patients with che-
moresistant disease but can result in a high rate of negative MRD, sometimes
with no remaining normal B cells, which could theoretically lead to an insuf-
cient target level for adequate CAR-T cell expansion and persistence. In a recent
clinical trial (Baruchel etal. 2020), this treatment was associated with a dimin-
ished EFS.Inotuzumab ozogamicin is also not recommended prior to the use of
anti-CD22 CAR-T cells.
References
Baruchel A, Krueger J, Balduzzi A. Tisagenlecleucel for pediatric/young adult patients with
relapsed/refractory b-cell acute lymphoblastic leukemia: preliminary report of B2001X focus-
ing on prior exposure to blinatumomab and inotuzumab. EHA Library. 2020;294938:S118.
Grupp SA, Maude SL, Rives S, etal. Updated analysis of the efcacy and safety of tisagenle-
cleucel in pediatric and young adult patients with relapsed/refractory (r/r) acute lymphoblastic
leukemia. Blood. 2018;132(Suppl 1):895.
Pillai V, Muralidharan K, Meng W, et al. CAR-T cell therapy is effective for CD19-dim
B-lymphoblastic leukemia but is impacted by prior blinatumomab therapy. Blood Adv.
2019;3:3539–49.
Taraseviciute A, Steinberg SM, Myers RM, et al. Pre-CAR blinatumomab is associated with
increased post-CD19 CAR relapse and decreased event free survival. Blood. 2020;136(Suppl
1):13–4.
Key Points
• The aim of optimal bridging therapy is not to obtain the lowest residual
disease possible but only to reduce or stabilize the tumour burden, bringing
the patient to CAR-T cell infusion in good clinical condition.
• There is no “one size ts all” in this area. Deep knowledge of disease biol-
ogy, emerging targets, previous sensitivity to treatment, and the tolerance
history of the patient are needed.
21 Bridging toCAR-T Cells inChildren, Adolescents, andYoung Adults withALL

118
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
A. Baruchel

119
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_22
C. Thieblemont (*)
Université de Paris, Paris, France
Hôpital Saint-Louis, Service d’Hémato-oncologie, DMU DHI, Assistance Publique des
Hôpitaux de Paris, Paris, France
e-mail: [email protected]
P. Borchmann
Department I of Internal Medicine, University Hospital of Cologne, Cologne, Germany
e-mail: peter[email protected]
22
Bridging Chemotherapy:
Relapsed/Refractory Aggressive
B-Cell Lymphoma
CatherineThieblemont andPeterBorchmann
To Bridge or Not toBridge inRelapsed andRefractory (R/R)
Aggressive B-Cell Lymphoma?
The time interval between leukapheresis and CAR-T cell infusion is critical for
many patients with R/R B-cell lymphoma and symptomatic disease that could be
fatal if left untreated during the cell manufacturing period. Often, oncologists
address this dilemma with bridging therapy (BT), which may include steroids, che-
motherapy, targeted therapy, or radiation therapy.
However, the R/R DLBCL patients included in the ZUMA-1 trial that led to axi-
cel approval were not allowed to receive BT other than dexamethasone (Neelapu
etal. 2017). In contrast, the JULIET and TRANSCEND trials leading to approval
of tisa-cel and liso-cel, respectively, allowed various treatments, including systemic
therapy, radiation therapy, or both (Schuster etal. 2019; Locke etal. 2019; Abramson
etal. 2020). In these latest trials, bridging therapy was used in 159 (59%) of 269
patients in TRANSCEND and 92% of patients in JULIET at the investigator’s dis-
cretion. Importantly, patients receiving BT are likely to have more aggressive dis-
ease and, therefore, are more likely to have other risk factors for increased rates of
cytokine release syndrome, neurological events, or both, such as an increased serum
lactate dehydrogenase (LDH) level, increased sum of product diameter or higher

120
total metabolic tumour burden (TMTV) before lymphodepleting chemotherapy, and
increased baseline C-reactive protein level.
Up to 87% of patients treated with axi-cel or tisa-cel in real-world settings
required BT (Nastoupil et al. 2020; Vercellino et al. 2020). However, in most
patients, bridging therapy did not result in a lower tumour burden at the time of
CAR-T cell reinfusion (Locke etal. 2019).
Which Bridging Therapy?
The time between leukapheresis and infusion may vary between the USA and
Europe. In France, this duration of time was described to be approximately 50days.
The median number of treatment cycles during the bridge was two cycles (range,
1–4; IQR, 1–2). Bridging therapy consisted of various immunochemotherapy or
chemotherapy regimens described as high-intensity BT and low-dose BT, including
chemo-free regimens. Because of lymphoma progression, 31% received more than
1 line of bridging therapy. Table22.1 describes different BT options.
The choice will be based on
1. Evaluation of the tumour mass and growth kinetics, including the LDH level,
sum of product diameter, and/or TMTV;
2. The biology of the disease, including cell of origin;
3. The type of prior lines and refractoriness or sensitivity; and
4. Patient characteristics, including comorbidities and resilience.
Table 22.1 Possible bridging therapies
No treatment: asymptomatic disease without clinically relevant tumour mass or growth
Low-intensity treatment: low disease burden (For off-label drug use, please check the local
requirements)
• Rituximab-dexamethasone
• Brentuximab vedotin
• Lenalidomide
• Radiotherapy
• Single agent chemotherapy, e.g., etoposide, gemcitabine, pixantrone
High-intensity treatment: aggressive disease
• Ifosfamide-VP16 with or without rituximab
• ICE (ifosfamide-carboplatinum-etoposide) with or without brentuximab vedotin or rituximab
• GEMOX (gemcitabine-oxaliplatin) with or without rituximab
• Polatuzumab-bendamustine-rituximab
Very high-intensity treatment for rapidly progressing disease
• High-dose melphalan with autologous stem cell support
• Hyperfractionated cyclophosphamide in combination regimens, e.g., hyperCVAD
C. Thieblemont and P. Borchmann

121
Response Rate toBridging Therapy andSubsequent CAR-T
Cell Therapy
Across the clinical trials, objective responses were achieved across all subgroups,
including in patients receiving bridging therapy (Locke et al. 2019). Durable
responses were also seen in patients who received BT (Locke etal. 2019). However,
in real life, early relapses were associated with high-intensity BT (Vercellino etal.
2020), correlated with a higher tumour burden and more rapidly progressive symp-
tomatic disease at the time of selection. Similarly, Nastoupil and coworkers showed
that among the patients who received axi-cel infusion, bridging therapy was not
associated with OS but may have negatively affected PFS, particularly among those
who received systemic BT.Intriguingly, patients who received bridging radiother-
apy had superior PFS vs. patients who were bridged with systemic therapy, despite
comparable baseline characteristics (Nastoupil etal. 2020).
References
Abramson JS, Palomba LM, Gordon LI, etal. Lisocabtagene maraleucel for patients with relapsed
or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless
design study. Lancet. 2020;396(10254):839–52.
Locke F, Ghobadi A, Jacobson C, etal. Long-term safety and activity of axicabtagene ciloleucel in
refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1-2 trial. Lancet
Oncol. 2019;20(1):31–42.
Key Points
The decisions of whether to administer bridging therapy and which
bridging therapy to choose require consideration of the following factors:
• Evaluation of the lymphoma growth kinetics based on measurement of
the tumour volume at the time of apheresis, ideally measured by total met-
abolic tumour volume on an 18-FDG PET scan or bulk disease (>7.5cm)
or widespread disease (Ann Arbor III or IV) evaluated on a CT scan, and
elevation of LDH levels above the upper normal value (UNV).
• Integration of the biology of the tumour with the possibility of choosing
targeted therapies, such as therapy with a monoclonal antibody (anti-
CD30, anti- CD20), BTK inhibitor or lenalidomide.
• Close monitoring of the patient, who may not be completely under your
control: a close collaboration and numerous contacts (at least 1/week) with
the referring centre is mandatory.
• All treatments can be used based on the growth kinetics and biology of
the tumour, individual patient characteristics, and prior lines, with one
goal: controlling the disease up to the time of CAR-T cell reinfusion.
22 Bridging Chemotherapy: Relapsed/Refractory Aggressive B-Cell Lymphoma

122
Open Access
This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
Nastoupil LJ, Jain MD, Feng L, etal. Standard-of-care axicabtagene ciloleucel for relapsed or
refractory large B-cell lymphoma: results from the US Lymphoma CAR-T Consortium. J Clin
Oncol. 2020;38(27):3119–28.
Neelapu S, Locke F, Bartlett N, etal. Axicabtagene ciloleucel CAR-T cell therapy in refractory
large B-cell lymphoma. N Engl J Med. 2017;377(26):2531–44.
Schuster S, Bishop M, Tam C, etal. JULIET Investigators. Tisagenlecleucel in adult relapsed or
refractory diffuse large B-cell lymphoma. N Engl J Med. 2019;380(1):45–56.
Vercellino L, Di Blasi R, Kanoun S, etal. Predictive factors of early progression after CAR-T cell
therapy in relapsed/refractory diffuse large B-cell lymphoma. Blood Adv. 2020;4(22):5607–15.
C. Thieblemont and P. Borchmann

123
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_23
N. Gagelmann · N. Kröger (*)
Department of Stem Cell Transplantation, University Medical Center Hamburg-Eppendorf,
Hamburg, Germany
e-mail: [email protected]; n.kroe[email protected]
J. Gribben
Bart’s Cancer Institute, Queen Mary University of London, London, UK
e-mail: [email protected]
23
Bridging Chemotherapy: Follicular
Lymphoma, Mantle Cell Lymphoma,
andCLL
NicoGagelmann, JohnGribben
, andNicolausKröger
While lisocabtagene maraleucel (liso-cel) is the only product that is specically
approved to treat grade 3B follicular lymphoma (FL), specically in aggressive
lymphoma indications, to date, axi-cel is the rst and only approved CAR-T product
for indolent NHL.
ZUMA-5 is a phase 2 study of axi-cel in patients with indolent NHL (including
FL and marginal zone lymphoma (MZL)) treated with two or more prior lines of
systemic therapy, with prior exposure to both an alkylating agent and anti-CD20
therapy (Jacobson etal. 2021). Of the 104 patients evaluable for efcacy, the ORR
was 92% and the CR was 76%. For the 84 patients with FL, the ORR was 95% (CR
80%), and for the 20 patients with MZL, the ORR was 85% (CR 60%). No differ-
ences between prior treatments were noted, while to date, specic analyses accord-
ing to bridging have not yet been presented. In the ELARA trial on tisa-cel in FL,
97 patients received treatment (median follow-up time, 10.6 months) (Schuster
etal. 2021). The median number of prior therapies was 4 (range, 2–13); 78% of
patients were refractory to their last treatment (76% to any ≥2 prior regimens) and
60% progressed within 2years of initial anti-CD20-containing treatment. The CR
rate was 66%, and the ORR was 86%, which was comparable among key sub-
groups, including bridging.
However, notably, indolent NHL is a chronic disease that can relapse after years
of remission. Although the rates of continued CR and PFS at 12months reported in
ZUMA-5 are encouraging, a longer follow-up time is needed to identify patients
who benet the most from certain treatment sequences.

124
Among relapsed or refractory mantle cell lymphoma patients receiving KTE-
X19 CAR-T therapy (Wang etal. 2020), a total of 25 patients (37% of the total
cohort) received bridging therapy with ibrutinib (14 patients), acalabrutinib (5),
dexamethasone (12), or methylprednisolone (2). The majority of the patients who
had assessments both before and after bridging therapy showed an increase in the
median tumour burden after the receipt of bridging therapy. Response rates were
similar regardless of exposure to bridging therapy, but ongoing responses seemed to
be higher in patients without bridging therapy (67% vs. 38%).
With regard to chronic lymphocytic leukaemia, the TRANSCEND CLL 004
study of liso-cel included patients with standard or high-risk features treated with
≥3 or≥2 prior therapies (Siddiqi etal. 2021), respectively, including Bruton kinase
inhibitors. A total of 17 patients (74%) received bridging therapy during liso-cel
manufacturing, and response rates were consistent, with 82% and 45% achieving
overall and complete responses, respectively. Safety and efcacy were similar
between treatment groups. Another small study even suggested the feasibility of
concurrent ibrutinib with CD19 CAR-T therapy (Gauthier etal. 2020), but the pop-
ulation overall is still limited, and studies are ongoing.
References
Gauthier J, Hirayama AV, Purushe J, etal. Feasibility and efcacy of CD19-targeted CAR-T cells
with concurrent ibrutinib for CLL after ibrutinib failure. Blood. 2020;135:1650–60.
Jacobson CA, Chavez JC, Sehgal AR, etal. Outcomes in ZUMA-5 with axicabtagene ciloleucel
in patients with relapsed/refractory indolent non-Hodgkin lymphoma who had the high-risk
feature of early progression after rst chemoimmunotherapy. Abstract #S213. Presented at the
EHA2021 Virtual Congress 2021 Jun 9.
Schuster SJ, Dickinson MJ, Dreyling MH, etal. Efcacy and safety of tisagenlecleucel (Tisa-cel)
in adult patients (Pts) with relapsed/refractory follicular lymphoma (r/r FL): primary analysis
of the phase 2 Elara trial. J Clin Oncol. 2021;39:7508.
Siddiqi T, Soumerai JD, Dorritie KA, etal. Phase 1 TRANSCEND CLL 004 study of lisocabta-
gene maraleucel in patients with relapsed/refractory CLL or SLL.Blood. 2021;
Wang M, Munoz J, Goy A, etal. KTE-X19 CAR-T cell therapy in relapsed or refractory mantle-
cell lymphoma. N Engl J Med. 2020;382:1331–42.
Key Points
• Limited evidence on the role of bridging therapy in FL and indolent
lymphoma.
• Systemic therapy led to worse outcomes across lymphoma types, but the
reasons are elusive.
• Bendamustine should be avoided whenever possible.
• The association between tumour volume before and after bridging therapy
and the overall response after CAR-T cell therapy is still unclear in mantle
cell lymphoma.
• Bridging therapy in CLL with BTKi seems feasible.
N. Gagelmann et al.

125
Open Access
This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
23 Bridging Chemotherapy: Follicular Lymphoma, Mantle Cell Lymphoma, andCLL

127
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_24
S. Manier (*)
Department of Hematology, Lille University, CHU Lille, Lille, France
e-mail: [email protected]
A. Jurczyszyn
Plasma Cell Dyscrasia Center, Department of Hematology, Jagiellonian University Medical
College, Krakow, Poland
e-mail: mmjurczy@cyf-kr.edu.pl
D. H. Vesole
Hackensack Meridian School of Medicine, Hackensack, NJ, USA
e-mail: David.V[email protected]g
24
Bridging Chemotherapy: Multiple
Myeloma
SalomonManier, ArturJurczyszyn, andDavidH.Vesole
Should All MM Patients Receive Bridging Therapy?
In the phase 2 KarMMa study, 88% of the patients received bridging therapy with
only a 5% response (Munshi etal. 2021). In the CARTITUDE 1 trial, 75% of the
patients received bridging therapy, with a reduction in tumour burden observed in
34% of the patients prior to cilta-cel infusion, but no patients achieved a CR or bet-
ter while on bridging therapy (Madduri etal. 2019). Bridging therapy is recom-
mended for virtually all patients. An exception can be discussed for patients with
slowly progressive disease, who may not need to receive bridging therapy after
leukapheresis; however, this strategy exposes them to a risk of rapid progression
later during the manufacturing period. In the future, with allogeneic CAR-T cells,
bridging therapy will likely not be necessary because the time between patient
inclusion and CAR-T cell infusion is much reduced.

128
Timeframe toUse Bridging Treatments
Bridging therapy can be started immediately after leukapheresis. Most clinical trials
do not permit the use of any bridging therapy within 2weeks prior to lymphodeple-
tion to allow for haematologic recovery and to prevent any interaction between the
drugs and the CAR-T cells (Munshi etal. 2021; Madduri etal. 2019).
Choice ofTreatment
Several clinical trials allow only agents to which the patients have been previously
exposed. However, this strategy can limit the efcacy of bridging therapy if patients
are refractory to their previous treatments. Therefore, bridging therapies are typi-
cally personalized to each patient according to previous lines of treatment, disease
characteristics, and pre-existing toxicities. All treatments can be considered for
bridging therapy, including proteasome inhibitors, immunomodulatory drugs, anti-
CD38 antibodies, targeted therapies, and conventional chemotherapies, with the
exception of anti-BCMA targeting therapies in the case of BCMA-targeting CAR-T
cells to avoid saturation of antigens. The risk of prolonged cytopenia and the risk of
infection should also be taken into account when considering conventional chemo-
therapies. Involved eld radiation therapy has been safely used during bridging
(Manjunath etal. 2020).
References
Madduri D, Usmani SZ, Jagannath S, etal. Results from CARTITUDE-1: a phase 1b/2 study of
JNJ-4528, a CAR-T cell therapy directed against B-cell maturation antigen (BCMA), in patients
with relapsed and/or refractory multiple myeloma (R/R MM). Blood. 2019;134(Suppl 1):577.
Manjunath SH, Cohen AD, Arscott WT, Maity A, Plastaras JP, Paydar I.Is Bridging Radiation
(RT) Safe with B Cell Maturation Antigen–targeting Chimeric Antigenic Receptor T Cells
(CART-BCMA) Therapy? ASTRO. 2020;1104
Munshi NC, Anderson LD Jr, Shah N, etal. Idecabtagene vicleucel in relapsed and refractory
multiple myeloma. N Engl J Med. 2021;384(8):705–16.
Key Points
• Virtually all patients should receive bridging therapy to prevent rapid pro-
gression of the disease during the manufacturing period.
• All treatments can be used with the exception of anti-BCMA therapies in
the case of BCMA-targeting CAR-T cells.
S. Manier et al.

129
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
24 Bridging Chemotherapy: Multiple Myeloma

131
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_25
M. Mohty (*)
Sorbonne University, Saint-Antoine Hospital, Paris, France
e-mail: [email protected]
M. C. Minnema
Department of Hematology, University Medical Center Utrecht, University Utrecht,
Utrecht, The Netherlands
e-mail: [email protected]
25
Lymphodepleting Conditioning
Regimens
MohamadMohty andMoniqueC.Minnema
Lymphodepleting conditioning regimens are essential for the success of CAR-T cell
treatment. Their importance in the proliferation and persistence of CAR-T cells has
become clearer in the recent years. The suggested mechanisms are described in
Table25.1 and include the effects on immune cells and cytokines, creating an envi-
ronment for optimal functioning and increasing the peak of expansion of the infused
CAR-T cells (Neelapu 2019).
The addition of udarabine to cyclophosphamide has been important in increas-
ing the efcacy of CAR-T cell treatment and is currently the most commonly used
combination (Turtle etal. 2016). In the applied conditioning regimens, the dosing of
udarabine is relatively consistent, with the use of 25–30mg/m
2
, given on 3 sequen-
tial days, but the dosing of cyclophosphamide differs in days and intensity. A “higher
intensity” cyclophosphamide dosing regimen seems to be preferred (Hirayama etal.
2019). However, even with the best lymphodepletion regimen, some patients fail to
develop a favourable cytokine prole, suggesting that the host biological response
to lymphodepletion chemotherapy is important (Hirayama etal. 2019). Most condi-
tioning regimens can be given on an outpatient basis.
In Hodgkin lymphoma treated with anti-CD30 CAR-T cells, bendamustine has
been used as conditioning regimen, but in this disease, the addition of udarabine to
the regimen has also been shown to increase antitumour responses. Whether an even
more intensive regimen is needed in solid tumours is currently unknown.

132
The timing of the conditioning regimen is typically within a week before the
planned infusion, with a minimum of 2 resting days to avoid a negative impact of
chemotherapy on the infused cells. If, after the start of the conditioning regimen, the
patient cannot receive CAR-T cells, most protocols allow a waiting time of
2–4weeks before a new conditioning regimen must be started. In other protocols,
conditioning regimens are not given if the absolute lymphocyte count is below 200
cells/μL.
The negative effects of conditioning regimens include pancytopenia and pro-
longed immune suppression and add to the enhanced risk of (viral) infections seen
after CAR-T cell treatment. In addition, udarabine can induce fever, neurotoxicity,
cyclophosphamide haemorrhagic cystitis, and pericarditis, and both drugs may
increase the risk of secondary malignancies.
References
Hirayama AV, Gauthier J, Hay KA, et al. The response to lymphodepletion impacts PFS in
patients with aggressive non-Hodgkin lymphoma treated with CD19 CAR-T cells. Blood.
2019;133:1876–87.
Neelapu SS.CAR-T efcacy: is conditioning the key? Blood. 2019;133:1799–800.
Turtle CJ, Hana L-A, Berger C, etal. Immunotherapy of non-Hodgkin’s lymphoma with a dened
ratio of CD8+ and CD4+ CD19-specic chimeric antigen receptor–modied T cells. Sci Transl
Med. 2016;8:1–12.
Table 25.1 Adapted from Neelapu (2019)
Effects of a conditioning regimen
Lymphodepletion Lowers total NK, B, and T cells
Fewer anti-CAR-T cell immune responses Reduces anti-transgene immune reactions
Eradication of immune suppressor cells Tregs and MDSCs
Modulation of tumour suppressive effects Lowers IDO expression, increases levels of
costimulatory molecules
Elimination of homeostatic cytokine sink Increases IL-2, IL-7, IL-15, and MCP-1
expression levels
Increased expansion, function, and
persistence of CAR-T cells
Better and durable tumour responses
Tregs regulatory T cells, MDSCs myeloid-derived suppressor cells, IDO indoleamine deoxygen-
ase, MCP-1 monocyte chemoattractant protein-1
Key Points
• An effective lymphodepleting regimen increases the proliferation and per-
sistence of CAR-T cells.
• Fludarabine seems essential and is typically used with
cyclophosphamide.
M. Mohty and M. C. Minnema

133
Open Access
This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
25 Lymphodepleting Conditioning Regimens

135
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_26
F. A. Ayuketang
Department of Stem Cell Transplantation, University Medical Center Hamburg,
Hamburg, Germany
e-mail: [email protected]
U. Jäger (
*)
Medical University of Vienna, Department of Medicine I, Division of Hematology and
Hemostaseology, Vienna, Austria
e-mail: [email protected]
26
Management ofCytokine Release
Syndrome (CRS) andHLH
FrancisAyukAyuketang andUlrichJäger
Cytokine Release Syndrome (CRS)
Definition andOccurrence
Cytokine release syndrome (CRS) is caused by a rapid and mild to massive release
of cytokines from immune cells involved in immune reactions, particularly after
immunotherapy. The frequency and severity of CRS after CAR-T cell therapy varies
between products (any grade: 37–93%, G3/4: 1–23%) (Neelapu etal. 2017; Schuster
etal. 2019; Abramson etal. 2020).
Diagnosis
Clinical Symptoms, Laboratory Diagnosis, Differential Diagnosis,
andPredictive Factors
CRS usually manifests with fever preceding or accompanied by general symptoms,
such as malaise, headache, arthralgia, anorexia, rigours, and fatigue, and can rapidly
progress to hypoxia, tachypnoea, tachycardia, hypotension, arrhythmia, culminat-
ing in shock cardiorespiratory organ dysfunction, and failure.
Although the diagnosis of CRS cannot be established or ruled out by laboratory
diagnostics, they can be used to monitor organ dysfunction. CRS symptoms and
laboratory ndings closely mimic infection; therefore, infectious workup and

136
treatment are of primary importance. Other relevant differential diagnoses include
tumour lysis and progression of the underlying malignancy.
Prediction of CRS in an individual patient is not yes possible. However, some
factors, such as high tumour burden and CAR-T cell dose, seem to be associated
with a higher risk of CRS.
Management
Patients receiving CAR-T cells should be monitored continuously or at regular
intervals for cardiovascular function and temperature. The rst sign of CRS is usu-
ally fever. Mild CRS (G1) can be managed conservatively. All higher grades require
intensive monitoring and intervention. Early use of tocilizumab and, in some cases,
steroids is now recommended (Table26.1) (Yakoub-Agha etal. 2020).
CRS Parameter
Grad 1
Grad 2
Grad 3 Grad 4
Fever
Hypotension
Hypoxia
Fever 38 C (not attributable to any other cause). In patients who have CRS then receive antipyretics or
anticytokine therapy such as tocilizumab or steriods, fever is no longer required to grade subsequent CRS
severity. In this case, CRS grading is driven by hypotension and/or hypoxia.
None
None
Not requiring
vasopressors
Requiring low-flow
oxygen (delivered at
6l/min)
Requiring a vasopressor with
or
without vasopressin
Requiring high-flow oxygen
(delivered at > 6l/min)
Requiring multiple
vasopressors
(excluding vasopressin)
Requiring positive
pressure (e.g. CPAP,
BiPA P, intubation and
mechanical ventilation)
Table 26.1 Scoring of CRS (adapted from Yakoub-Agha et al. 2020)
GRADE 1
GRADE 2
GRADE 3 GRADE 4
Alert your local ICU
Start preemptive broad-spectrum antibiotics and sympomatic measures
a
CRS treatmemt (outside clinical trials)
Toci IV 8 mg / kg (max = 800 mg) to be done in the hematology unit before tranfer to ICU
If deterioration
If deterioration
Dexamethasone IV 10 mg/6h
for 1-3 days
• Dexamethasone IV 10mg/6h
for 1-3 days
Dexamethasone IV 20 mg/6h
for 3 days, progressive
tapering within 3-7 days
In the absence of improvement
within 3 days and in the absence
of other differential diagnosis
In the absence of improvement at H+12, repeat TOCILIZUMAB IV 8 mg/kg (Max = 800 mg)
If absence of improvement, persistence of symptoms
Consider Toci IV 8 mg / kg
(max = 800 mg)
• Switch to Methylprednisolone IV
1000mg/d for 3 days then 250mg x
2/d for 2 days, 125mg x 2/d for 2
days, 60mg x 2/d for 2 days
• Consider repeating Toci (maximum
1 additional dose) in the asence of
ICANS
• Dexamethasone IV 20 mg/6h
for 1-3 days
Fig. 26.1 Management of CRS—Modied according to EBMT recommendations
F. A. Ayuketang and U. Jäger

137
Monitoring: Patients with CRS 1 can be monitored on the regular ward or
Intermediate Care ward, starting from G2, and admission to an ICU should be
considered.
Supportive therapy consists of uids and antipyretics. The use of vasopressors
automatically marks higher grade CRS.
Anti-Cytokines
Tocilizumab is EMA and FDA approved for the treatment of CRS.Prophylactic,
preemptive or risk-adapted use may reduce the risk of severe CRS without attenuat-
ing antitumour efcacy.
(Locke etal. 2017; Caimi etal. 2020; Gardner etal. 2019; Kadauke etal. 2021).
Clinical trial data on the use of siltuximab and anakinra are still lacking.
Steroids
In contrast to initial clinical studies, short courses of steroids do not seem to have
detrimental effects on CAR-T cell expansion and survival or clinical outcome.
Antibiotics
Because CRS cannot be decisively differentiated from infection, most centres
administer antibiotic treatment in cases of neutropenic fever. However, the use of
growth factors during the rst few weeks should be restricted. GM-CSF is to be
avoided.
sHLH/MAS
Secondary or reactive haemophagocytic lymphohistiocytosis (sHLH) is a life-
threatening hyperinammation syndrome that occurs in the context of allo-HCT,
haematological malignancies, infection, and rheumatic or autoimmune disease and
is characterized by hyperactive macrophages and lymphocytes, haemophagocyto-
sis, and multiorgan damage (Carter etal. 2019; Neelapu etal. 2018; Sandler etal.
2020). The proposed diagnostic criteria are summarized in Table26.2. Management
of sHLH generally follows similar algorithms as that for severe CRS.In refractory
patients, treatment may follow the management framework proposed by Mehta
etal. (2020), with a key role for anakinra.
26 Management ofCytokine Release Syndrome (CRS) andHLH

138
References
Abramson JS, Palomba ML, Gordon LI, etal. Lisocabtagene maraleucel for patients with relapsed
or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless
design study. Lancet. 2020;396(10254):839–52.
Caimi PF, Sharma A, Rojas P, etal. CAR-T therapy for lymphoma with prophylactic tocilizumab:
decreased rates of severe cytokine release syndrome without excessive neurologic toxicity.
Blood. 2020;136:30–1. https://ash.confex.com/ash/2020/webprogram/Paper143114.html
Carter SJ, Tattersall RS, Ramanan AV.Macrophage activation syndrome in adults: recent advances
in pathophysiology, diagnosis and treatment. Rheumatology (Oxford). 2019;58(1):5–17.
Gardner RA, Ceppi F, Rivers J, etal. Preemptive mitigation of CD19 CAR-T cell cytokine release
syndrome without attenuation of antileukemic efcacy. Blood. 2019;134(24):2149–58.
Kadauke S, Myers RM, Li Y, etal. Risk-adapted preemptive tocilizumab to prevent severe cytokine
release syndrome after CTL019 for pediatric B-cell acute lymphoblastic leukemia: a prospec-
tive clinical trial. J Clin Oncol. 2021;39(8):920–30.
Diagnostik criteria for CAR-T cell mediated sHLA/MAS
(Neelapu et al., 2018)
Adult HScore (Fardet et al., 2014): Respective scores are in brackets
Produces a probability outcome; scores >169 are 93% sensitive and
86% specific for HLH
Clinical Clinical
Grade
3 pulmonary oedema* Fever
Hepatomegaly /
Immunosuppression
<38.4 (0); 38.4 39.4 (33); >39.4 (49)
Neither (0); either hepatomegaly or
splenomegaly (23); both (38)
No (0); yes (18)
Laboratory
Ferritin, ng/mL
Cytopenias >2 lineages
<2000 (0); 2000-6000 (35); >6000
(50)
One lineage (0), two lineages (24), or
three lineages (34)
Hypertriglyceridaemia,
mmol
<1.5 (0); 1.5-4 (44); >4 (64)
Haemophagocytosis
No (0), yes (35)
Labotarory
Peak ferritin 10.000ng/ml during CRS and any 2 of the
following
Grade
3 increase in serum bilirubin, aspartate
aminotransferase, or alanine aminotransferase levels*
Grade
3 oliguria or increase in serum creatinine levels*
Presence of haemophagocytosis in bone marrow or
organs based on histopathological assessment of cell
morphology and/or CD68 immunohistochemistry
*According to Common Te rminology Criteria for Adverse
Events (CTCAE) Version 4.0
Liver function tests, IU/L
Hypofibrinogenaemia, g/L
AST<30 (0); >30 (19)
>2.5 (0); <2.5 (30)
Table 26.2 Diagnostic criteria for HLA (adapted from Neelapu etal. 2018)
Key Points
• Cytokine release syndrome is a frequent complication. However, severe
CRS is rare if management is proactive.
• sHLH/MAS is a rare but severe complication that requires prompt recogni-
tion and intervention.
F. A. Ayuketang and U. Jäger

139
Locke FL, Neelapu SS, Bartlett NL, etal. Preliminary results of prophylactic Tocilizumab after
axicabtagene ciloleucel (axi-cel; KTE-C19) treatment for patients with refractory, aggressive
non-Hodgkin lymphoma (NHL). Blood. 2017;130(Supplement 1):1547.
Mehta P, Cron RQ, Hartwell J, Manson JJ, Tattersall RS.Silencing the cytokine storm: the use of
intravenous anakinra in haemophagocytic lymphohistiocytosis or macrophage activation syn-
drome. Lancet Rheumatol. 2020;2(6):e358–67.
Neelapu SS, Locke FL, Bartlett NL, etal. Axicabtagene Ciloleucel CAR-T cell therapy in refrac-
tory large B-cell lymphoma. N Engl J Med. 2017;377(26):2531–44.
Neelapu SS, Tummala S, Kebriaei P, etal. Chimeric antigen receptor T-cell therapy– assessment
and management of toxicities. Nat Rev Clin Oncol. 2018;15(1):47–62.
Sandler RD, Carter S, Kaur H, Francis S, Tattersall RS, Snowden JA.Haemophagocytic lympho-
histiocytosis (HLH) following allogeneic haematopoietic stem cell transplantation (HSCT)-
time to reappraise with modern diagnostic and treatment strategies? Bone Marrow Transplant.
2020;55(2):307–16.
Schuster SJ, Bishop MR, Tam CS, etal. Tisagenlecleucel in adult relapsed or refractory diffuse
large B-cell lymphoma. N Engl J Med. 2019;380(1):45–56.
Yakoub-Agha I, Chabannon C, Bader P, etal. Management of adults and children undergoing chi-
meric antigen receptor T-cell therapy: best practice recommendations of the European Society
for Blood and Marrow Transplantation (EBMT) and the joint accreditation committee of ISCT
and EBMT (JACIE). Haematologica. 2020;105(2):297–316.
Open Access
This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
26 Management ofCytokine Release Syndrome (CRS) andHLH

141
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_27
J. H. Rees (*)
National Hospital for Neurology and Neurosurgery, University College London Hospitals
NHS Trust, London, UK
UCL Institute of Neurology, London, UK
e-mail: jeremy[email protected]
27
Management ofImmune Effector
Cell- Associated Neurotoxicity Syndrome
(ICANS)
JeremyH.Rees
A common and challenging side effect associated with CAR-T cell therapy is
immune cell-associated neurotoxicity syndrome (ICANS), which occurs in 20–60%
of patients, of whom 12–30% have severe (≥ grade 3) symptoms.
The underlying mechanism driving the syndrome is not fully understood, but
there is evidence for the release of inammatory cytokines secreted by macrophages
and monocytes, increasing vascular permeability and endothelial activation and
leading to blood–brain barrier breakdown. ICANS is not thought to be directly
mediated by CAR-T cells themselves.
Risk factors for ICANS include high disease burden, older age, and the specic
CAR-T product.
The onset of ICANS occurs (on average) approximately 5days following CAR-T
cell infusion and sometimes occurs concurrently with or shortly after cytokine
release syndrome (CRS). However, in approximately 10% of patients, ICANS pres-
ents more than 3weeks after CAR-T cell infusion.
Symptoms of ICANS are variable and can initially be vague. Patients experience
mild tremor and confusion, which can then proceed to agitation, seizures, and cere-
bral oedema. A prominent and early feature of ICANS is hesitancy of speech and
deterioration in handwriting, which can progress to aphasia with both expressive
and receptive components, whereby the patient is alert but mute. The most devastat-
ing consequence of ICANS is the occurrence of status epilepticus, fatal cerebral
oedema and occasionally intracerebral haemorrhage.
ICANS is a clinical diagnosis—brain MRI and CSF evaluation are rarely helpful
but can be used to rule out alternative diagnoses, e.g., CNS infection. The EEG

142
recording can be normal but can also demonstrate a pattern of variable abnormali-
ties, including nonconvulsive status epilepticus.
Most cases spontaneously resolve, often with supportive care and early interven-
tion with corticosteroid therapy.
All patients should be proactively monitored for ICANS twice daily to assess
subtle changes in cognition using the 10-point Immune Effector Cell Encephalopathy
(ICE) score (Table27.1), which evaluates orientation, attention, writing, and lan-
guage. This score is then integrated into an overall assessment of neurological func-
tion incorporating seizure activity, change in consciousness level, motor ndings,
and elevation in intracerebral pressure/cerebral oedema to obtain an ICANS grade.
The higher the ICE score is, the lower the ICANS grade. Any patient with an ICE
score less than 2 or with seizures is classied as severe (grade 3 or 4) and should be
transferred to intensive care. Factors associated with a higher risk of ≥ grade 3
ICANS include a higher disease burden, low platelet count, and the development of
early and severe CRS.
Management of ICANS is based on the severity of the score and the concurrence
of CRS.Management is supportive for grade 1 ICANS, and dexamethasone with
rapid taper is given for grade ≥2 ICANS.Suggested doses include 10–20mg intra-
venous dexamethasone every 6h for grades 2–3 and 1g IV methylprednisolone for
at least 3days for grade 4 until symptoms improve. Seizures are treated with leveti-
racetam and status epilepticus with benzodiazepines. We do not recommend the use
of prophylactic anti-epileptic drugs.
Other experimental approaches to the management of ICANS have been directed
at controlling the potency of the CAR itself. Several CAR constructs have been
designed with “suicide switches” or as “tunable CARs” by incorporating mecha-
nisms designed to turn off or downgrade the CAR in the event of severe toxicity. In
severe unresponsive cases, anakinra (IL-1 receptor antagonist) or chemotherapy to
kill the CAR-T cells have been used. However, most cases resolve and do not result
in residual neurocognitive damage (Tables 27.2 and 27.3).
Table 27.1 Immune Effector Cell Encephalopathy (ICE) Score
Immune Effector Cell Encephalopathy (ICE) Score
• Orientation: Orientation to year, month, city, hospital: 4 points
• Naming: Ability to name 3 objects (e.g., point to clock, pen, button): 3 points
• Following commands: Ability to follow simple commands (e.g., “show me 2 ngers” or
“close your eyes and stick out your tongue”): 1 point
• Writing: Ability to write a standard sentence (e.g., “our national bird is the bald eagle”): 1
point
• Attention: Ability to count backwards from 100 by 10: 1 point
J. H. Rees

143
Key Points
• ICANS is a common and usually reversible toxicity of CAR-T cell therapy,
occurring within a week of infusion, often after cytokine release syndrome.
• ICANS is a clinical diagnosis—common early symptoms include word
nding difculties, confusion, and impaired ne motor skills. Investigations
are rarely helpful except to rule out an alternative diagnosis, such as CNS
infection.
• Severe ICANS consists of seizures, coma, and cerebral oedema and
requires ITU care.
• Management of ICANS is largely supportive and depends on severity.
Corticosteroids are the mainstay of care for all but Grade I ICANS and
should be prescribed at high doses with a rapid taper.
• The prognosis is good, and the majority of patients fully recover without
any long-term sequelae.
Table 27.2 American Society
for Transplantation and
Cellular Therapy (ASTCT)
ICANS Consensus Grading
for Adults
(continued)
Overall ICANS Grade
Grade 1
Grade 2
Grade 3
Grade 4
ICE score
a
7–9
3–6
0–2
0 (patient is unarousable and unable to perform the ICE
assessment)
Depressed level of consciousness
b
Awakens spontaneously
Awakens to voice
Awakens only to tactile stimulus
Patient is unarousable or requires vigorous or repetitive
tactile stimuli to arouse. Stupor or coma
Seizure
N/A
N/A
Any clinical seizure focal or generalized that resolves
rapidly or nonconvulsive seizures on EEG that resolve
with intervention
Life-threatening prolonged seizure (>5min) or repetitive
clinical or electrical seizures without return to baseline in
between
27 Management ofImmune Eector Cell-Associated Neurotoxicity Syndrome (ICANS)

144
Table 27.3 Approach to management of ICANS
ICANS
Grade Management
Grade 1 Consider levetiracetam seizure prophylaxis (750mg BD)
Avoid medications that cause central nervous system depression
Seek neurology specialist consultation
Supportive care
Fundoscopic examination to assess for papilloedema
Brain MRI with contrast (brain CT if brain MRI is not feasible)
Consider diagnostic lumbar puncture with measurement of opening pressure where
possible, sending samples for culture and sensitivity, cytology, biochemistry, and
virology as a minimum
Consider spine MRI if the patient has focal peripheral neurological decits
Consider electroencephalogram (EEG)
Consider tocilizumab 8mg/kg but only if concurrent CRS
Twice daily neurocognitive assessment using the ICE score and ICANS grading
Grade 2 Investigations and supportive care as per grade 1
Consider dexamethasone at a high dose with rapid weaning
Consider transferring the patient to the intensive care unit (ICU)
Motor ndings
c
N/A
N/A
N/A
Deep focal motor weakness, such as hemiparesis or
paraparesis
Elevated ICP/cerebral oedema
N/A
N/A
Focal/local oedema on neuroimaging
d
Diffuse cerebral oedema on neuroimaging; decerebrate
or decorticate posturing; Sixth nerve palsy;
papilloedema; or Cushing’s triad
ICANS grade is determined by the most severe event (ICE
score, level of consciousness, seizure, motor ndings, raised
ICP/cerebral oedema) not attributable to any other cause; for
example, a patient with an ICE score of 3 who has a generalized
seizure is classied as having grade 3 ICANS
N/A indicates not applicable
a
A patient with an ICE score of 0 may be classied as having
grade 3 ICANS if awake with global aphasia, but a patient with
an ICE score of 0 may be classied as having grade 4 ICANS if
unarousable
b
Depressed level of consciousness should be attributable to no
other cause (e.g., no sedating medication)
c
Tremors and myoclonus associated with immune effector cell
therapies may be graded according to CTCAE v5.0, but they do
not inuence ICANS grading
d
Intracranial haemorrhage with or without associated oedema
is not considered a neurotoxicity feature and is excluded from
ICANS grading. It may be graded according to CTCAE v5.0
Table 27.2 (continued)
J. H. Rees

145
Further Reading
Neill L, Rees J, Roddie C.Neurotoxicity– CAR-T cell therapy: what the neurologist needs to
know. Pract Neurol. 2020;20:287–95. https://doi.org/10.1136/practneurol- 2020- 002550.
Rubin DB, Danish HH, Ali AB, etal. Neurological toxicities associated with chimeric antigen
receptor T-cell therapy. Brain. 2019;142:1334–48. https://doi.org/10.1093/brain/awz053.
Tallantyre EC, Evans NA, Parry-Jones J, etal. Neurological updates: neurological complications
of CAR-T therapy. J Neurol. 2021;268:1544–54. https://doi.org/10.1007/s00415- 020- 10237- 3.
Open Access
This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
ICANS
Grade Management
Grade 3 Investigations and supportive care as per grade 1
Administer dexamethasone 10–20mg IV every 6h or methylprednisolone
equivalent until improvement to grade 1 and then taper
Management of seizures with lorazepam 0.5mg IV or other benzodiazepines as
needed, followed by loading with levetiracetam or other anticonvulsants as required
If fundoscopy reveals stage 1 or 2 papilloedema with cerebrospinal uid (CSF)
opening pressure>20mmHg, seek urgent advice from neurologist
Consider repeat neuroimaging (CT or MRI) every 2–3days if the patient has
persistent grade≥3 ICANS
Grade 4 Investigations and supportive care as per grade 1
Transfer patient to intensive care unit (ICU); consider mechanical ventilation for
airway protection
Seizure management as per grade 3
For convulsive status epilepticus, seek urgent advice from neurologist
Administer methylprednisolone 1000mg/day for 3days, then taper at 250mg every
12hrs for 2days, then 125mg every 12hrs for 2days, then 60mg every 12hrs for
2days
For management of raised intracranial pressure, consider acetazolamide 1000mg
IV, followed by 250–1000mg IV every 12h; elevating the head of the bed;
hyperventilation; and hyperosmolar therapy with mannitol
Table 27.3 (continued)
27 Management ofImmune Eector Cell-Associated Neurotoxicity Syndrome (ICANS)

147
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_28
M. Topp
Medizinischen Klinik und Polklinik II, Universitätsklinikum Würzburg, Würzburg, Germany
e-mail: Topp_M@ukw.de
T. Feuchtinger (
*)
Department Pediatric Hematology, Oncology Hemostaseology and Stem Cell Transplantation,
Dr von Hauner Children’s Hospital, University Hospital, LMU, Munich, Germany
e-mail: T[email protected]
28
Management
ofHypogammaglobulinaemia
andB-Cell Aplasia
MaxTopp andTobiasFeuchtinger
The development and regulatory approval of CAR-T cell therapies targeting
B-lineage surface antigens (Maude etal. 2018), such as CD19 or CD22, represents
a major milestone in cancer immunotherapy. This treatment results in the depletion
of malignant and normal B cells and is associated with hypogammaglobulinaemia.
These on-target, off-tumour toxicities may result in an increased risk of infection.
Careful long-term follow-up assessment of patients receiving CAR-T cell therapy is
important. Management of these on-target, off-tumour effects should be well coor-
dinated between treatment and referring centres if the patient returns to local pro-
viders following therapy. Aims of this toxicity management:
• Prophylaxis of acute and chronic infections.
• Treatment of infections.
• Prevention of organ damage due to silent and/or chronic infections (e.g.,
bronchiectasis).
• Best-possible quality of life.
Monitoring B-cell aplasia provides information on two aspects of treatment.
First, B-cell aplasia is a sign of functional CAR persistence and often shows longer
persistence than direct detection of the CAR-T cells themselves. Monitoring B-cell
aplasia can guide decisions on the interval of monitoring of the malignant disease
(e.g., imaging, MRD) and subsequent allogeneic HSCT in cases of CAR-T cell
failure. Therefore, B-cell aplasia should be monitored together with remission

148
status and CAR persistence. Second, B-cell aplasia is a helpful parameter to guide
tapering or continuation of IgG substitution. The evidence level for the recommen-
dation concerning monitoring B-cell aplasia is based on expert opinion and may
vary in different patient age groups.
Because anti-CD19 and anti-CD22 CAR-T cells attack B-lineage precursors,
long-term hypogammaglobulinaemia or agammaglobulinaemia are commonly seen
in patients after CAR-T cell treatment. In paediatric centres, it has been the standard
of care to perform immunoglobulin replacement following CAR-T cell therapy
(Maude etal. 2014). However, there is no consensus regarding systematic IgG sup-
plementation in adults with plasma-cell aplasia and hypogammaglobulinaemia after
CAR-T cell therapy. Since persistent B-cell aplasia is associated with sinopulmo-
nary infections, notably with encapsulated bacteria (Fishman etal. 2019), intrave-
nous immunoglobulin replacement is performed in all patients with
hypogammaglobulinaemia plus recurrent or chronic infections, especially pneumo-
nia. After allogeneic HSCT plus CAR-T cell therapy, patients face an increased risk
of infection-related morbidity and hence may require intensied (perhaps lifelong)
IgG substitution regimens.
The duration of IgG substitution may be lifelong or last at least until recovery of
functional B cells and plasma cells. However, data regarding the efcacy of prophy-
lactic IgG replacement in CAR-T cell therapy recipients are limited (Hill et al.
2019), and current expert recommendations (Mahadeo etal. 2019, Yakoub-Agha
etal. 2020) are extrapolated from the data for individuals with primary immune
deciencies (Perez etal. 2017, Picard etal. 2018). The long-term follow-up data
from patients with Bruton agammaglobulinaemia provide a rationale to closely
monitor immunoglobulin levels and acute, chronic and especially silent infections
to prevent organ damage and maintain long-term quality of life. Individualized regi-
mens aim to maintain serum immunoglobulin levels above 400μg/l in adults and
age-adapted normal ranges for children. Intravenous immunoglobulins (IVIGs) are
usually given every 3–6weeks or subcutaneously weekly (SCIGs). IVIG doses start
at 0.4g/kg body weight and SCIG doses at 0.1–0.15g/kg body weight. Doses and
intervals are adapted due to infections and serum IgG levels. After reaching a steady
state, serum IgG levels should be controlled at least every 3months.
Studies are needed to establish evidence-based approaches for management of
B-cell aplasia and hypogammaglobulinaemia. Prophylactic immunoglobulin
administration in this context and strategies may differ by patient and CAR-T cell
product characteristics.
Key Points
• Long-term (potentially lifelong) monitoring of B-cell aplasia and IgG lev-
els is necessary after CD19/CD22 targeting CAR-T cell therapies.
• Maintain serum immunoglobulin levels at physiologic levels through regu-
lar intravenous or subcutaneous substitution.
• The evidence level of this recommendation is at the expert opinion level.
M. Topp and T. Feuchtinger

149
References
Fishman JA, Hogan JI, Maus MV. Inammatory and Infectious Syndromes Associated With
Cancer Immunotherapies. Clin Infect Dis. 2019;69(6):909–20.
Hill JA, Giralt S, Torgerson TR, Lazarus HM. CAR-T - and a side order of IgG, to go? -
Immunoglobulin replacement in patients receiving CAR-T cell therapy. Blood Rev.
2019;38:100596. e-pub ahead of print 2019/08/17
Mahadeo KM, Khazal SJ, Abdel-Azim H, Fitzgerald JC, Taraseviciute A, Bollard CM, et al.
Management guidelines for paediatric patients receiving chimeric antigen receptor T cell ther-
apy. Nat Rev Clin Oncol. 2019;16(1):45–63. e-pub ahead of print 2018/08/08
Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, etal. Chimeric antigen receptor T
cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–17. e-pub ahead
of print 2014/10/16
Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, etal. Tisagenlecleucel in chil-
dren and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439–48.
Perez EE, Orange JS, Bonilla F, Chinen J, Chinn IK, Dorsey M, etal. Update on the use of immu-
noglobulin in human disease: A review of evidence. J Allergy Clin Immunol. 2017;139(3S):S1–
S46. e-pub ahead of print 2017/01/04
Picard C, Bobby Gaspar H, Al-Herz W, Bousha A, Casanova JL, Chatila T, etal. International
Union of Immunological Societies: 2017 Primary immunodeciency diseases committee
report on inborn errors of immunity. J Clin Immunol. 2018;38(1):96–128. e-pub ahead of print
2017/12/12
Yakoub-Agha I, Chabannon C, Bader P, Basak GW, Bonig H, Ciceri F, etal. Management of adults
and children undergoing chimeric antigen receptor T-cell therapy: best practice recommenda-
tions of the European Society for Blood and Marrow Transplantation (EBMT) and the Joint
Accreditation Committee of ISCT and EBMT (JACIE). Haematologica. 2020;105(2):297–316.
e-pub ahead of print 2019/11/23
Open Access
This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
28 Management ofHypogammaglobulinaemia andB-Cell Aplasia

151
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_29
M. Subklewe (*)
Med. Klinik & Poliklinik III, LMU– Klinikum der Universität München, München, Germany
e-mail: [email protected]
R. Benjamin
King’s College Hospital, London, UK
e-mail: [email protected]
29
Management ofMyelotoxicity (Aplasia)
andInfectious Complications
MarionSubklewe andReubenBenjamin
Haematologic toxicity is the most common adverse event after CAR-T cell therapy,
with a cumulative 1-year incidence of 58% (CTCAE grade≥3) in the real-world
setting (Wudhikarn etal., Blood Advances 2020). It is characterized by a biphasic
temporal course and is often prolonged (Fried etal., Bone Marrow Transplant 2019,
Rejeski et al., Blood et al. 2021a, b, Fig. 29.1). In a report of Axi-Cel-treated
patients, only 30% demonstrated a neutrophil count >1×10
9
/L and 50% showed a
platelet count >50×10
9
/L at 30days following CAR-T cell treatment (Jain etal.,
Blood Advances 2020). In a long-term follow-up study of patients with ongoing CR
and an absence of MDS, 16% of patients experienced prolonged signicant cytope-
nias that lasted up to 22months after CAR-T cell treatment (Cordeiro etal., Biol
Blood Marrow Transplant 2020). These ndings highlight that cytopenia can pres-
ent long after lymphodepletion and the resolution of acute CRS.Risk factors include
severe CRS/ICANS, cytopenia prior to initiation of lymphodepleting chemother-
apy, and prior allogeneic stem cell transplantation (Jain et al., Blood Advances
2020, Fried et al., BMT, 2019). Importantly, cytopenia predisposes patients to
severe infectious complications, which are the most frequent cause of non-relapse
mortality (Nastoupil etal., JCO 2020).
The use of growth factors to manage early cytopenias after CAR-T cell therapy
remains controversial. Cytokine proles and mouse xenograft models have impli-
cated GM-CSF in the pathogenesis of CRS and neuroinammation (Sterner etal.,
Blood 2019). Accordingly, current recommendations discourage the use of GM-CSF
and to initiate G-CSF only after resolution of CRS and/or ICANS (Yakoub- Agha
et al., Haematologica 2020). However, in a report by Galli et al., prophylactic

152
G-CSF was used safely in 42 patients with grade 4 neutropenia on day 5 after
CAR-T cell infusion, with no increased risk of CRS or ICANS and no negative
impact on disease outcomes (Galli et al., Bone Marrow Transplantation 2020).
Further studies are needed to identify patients at high risk for prolonged neutropenia
and with an increased risk of infection that will benet from early G-CSF initiation.
The diagnostic workup for patients with prolonged cytopenia unresponsive to
G-CSF should include screening for haematinic deciency, viral infections (e.g.,
CMV, EBV, Hepatitis B/C, Parvovirus B19), concomitant myelosuppressive drugs
(e.g., co-trimoxazole), secondary haemophagocytic lymphohistiocytosis, and the
presence of disease in the bone marrow. In the event of severe bone marrow aplasia
unresponsive to G-CSF, autologous or allogeneic stem cell rescue may be consid-
ered where possible (Rejeski etal., BMC ID 2021a, b, Godel etal., Haemasphere
2021). Other options include anti-inammatory therapy (e.g., dexamethasone, anti-
IL- 6 blocking therapy) and thrombopoietin receptor agonists (e.g., eltrombopag)
(Fig.29.2).
Infections are another signicant complication of CAR-T therapy as a result of
prolonged neutropenia, long-term CD4 T cell lymphopenia, or B-cell aplasia (Hill
and Seo etal., Blood 2020a, b). Other risk factors associated with infections include
higher CRS grade and use of immunosuppressive agents, such as steroids, tocili-
zumab, and anakinra. The majority of infections occur early within the rst 28days,
with bacterial infections being the most common, followed by viral and fungal
infections. Late infections, especially with respiratory viruses, are also seen up to
90 days post CAR-T therapy (Cordeiro et al., BBMT 2020). Invasive fungal
6000
5000
4000
3000
2000
Abs. Neutrophil Count/µl
1000
0
-5 -4 -3 -2 -1
012345678910 11 12 13 14 W3 W4 W5 W6 W7 W8 M3 M4
MonthsWeeksDays
Baseline
Clinical Phenotypes of neutropenia after CAR T-cell transfusion
Quick Recovery Intermittent Recovery Aplastic
Fig. 29.1 Clinical Phenotypes of neutropenia (adapted from Rejeski etal., Blood 2021a, b)
M. Subklewe and R. Benjamin

153
infections, both yeasts and moulds, have been reported in 1–15% of CAR-T-treated
patients.
The use of antiviral, antifungal, and anti-pneumocystis (PCP) prophylaxis to
reduce the risk of infections post-CAR-T cell therapy is recommended; however,
there is no consensus on the optimal choice and duration of prophylaxis. Patients
typically receive acyclovir or valacyclovir, yeast or mould-active antifungal prophy-
laxis, and co-trimoxazole to prevent PCP.Patients considered “high risk” for mould
infections based on pre-infusion neutropenia, history of mould infection within
6 months, prior allo-SCT or underlying diagnosis of acute leukaemia as well as
those who have had post-infusion grade≥3 CRS/ICANS or prolonged treatment
with steroids or other immunosuppressants should be offered mould-active prophy-
laxis. In the absence of high-risk factors, yeast-active prophylaxis may be sufcient,
with pre-emptive monitoring for moulds recommended (Garner et al., J Fungi
2021). Antifungal prophylaxis is generally continued until recovery of the neutro-
phil count and cessation of immunosuppressants. PCP prophylaxis is typically
given for 6months or until the CD4 T cell count is >200 cells/μl. Monthly intrave-
nous immunoglobulins may be considered when there is persistent severe hypogam-
maglobulinaemia and a history of recurrent infections. Treatment of suspected/
conrmed infections post CAR-T therapy should follow Institutional guidelines and
is generally similar to the management of infections after stem cell transplantation.
The efcacy of vaccination following administration of CD19- or BCMA-targeted
CAR-T cells remains unknown, but patients do need to be considered for vaccina-
tion at 6months post-CAR-T therapy and guided by post-vaccine antibody titres
(Hill etal., Blood 2020a, b). Effective strategies to prevent and manage infectious
complications following CAR-T cell infusion are crucial in improving the outcomes
of this promising therapy.
ANC < 1000/µl
D7-10 post CAR-T
Filgrastim 0.5 Mio
IE/kgKG
until ANC > 1000/µl
“G-CSF
responsive”
“G-CSF
unresponsive”
• Vigilant outpatient monitoring of CBC
• consider bone marrow aspiration /
histology, if continuous G-CSF is
needed
• Posaconazol 300 mg p.o. or
Micafungin 50 mg i.v.
• Surveillance: Asp. AG, CMV/
EBV PCR weekly
• if ANC > 500/µl: watch and wait
• Pegylated G-CSF: e.g.
Lipegfilgrastim 6 mg s.c.
• Pulse-Dose Steriods:
Dexamethason 20mg x 4 days
• Anti-cytokine-Therapy: e.g.
Anakinra or Tocilizumab
• TPO-Agonist: e.g. Eltrombopag
50mg x 7-14 days
yes
yes
no
no
Infectious Prophylaxis:
Bone Marrow
Studies:
r/o Bone Marrow
infiltration?
Check for
autologous
back-up?
AutoSCT
individual
decision
regeneration?
Ultima Ratio:
Allogeneic SCT
Refractory
Extend Staging and
diagnostic studies. Consider
integrating CAR T-cell
monitoring => Initiate anti-
lymphoma/leukemia
therapy accoding to
patient-specific features
Warning:
Initiate therapy:
ANC < 1000/µl
ANC < 500/µl
Work-up:
• CBC
• reticulocytes, RPI
• r/o substrate
deficiency
• r/o drug induced
cytopenia
• bone marrow
aspiration /
histology
Rejeski et al, 2021, Godel et al, 2021
Hill and Seo et al, Blood Advances 2020
Fig. 29.2 Treatment algorithm for CD19 CAR-T cell-associated myelotoxicity
29 Management ofMyelotoxicity (Aplasia) andInfectious Complications

154
References
Cordeiro A, etal. Late events after treatment with CD19-targeted chimeric antigen receptor modi-
ed T cells. Biol Blood Marrow Transplant. 2020;26(1):26–33.
Fried S, etal. Early and late hematologic toxicity following CD19 CAR-T cells. Bone Marrow
Transplant. 2019;54(10):1643–50.
Galli E, Allain V, Blasi R, Bernad S, Vercellino L, Morin F, Moatti H, Caillat-Zucman S, Chevret
S, Thieblemont C.G-CSF does not worsen toxicities and efcacy of CAR-T cells in refractory/
relapsed B-cell lymphoma. Bone Marrow Transplant. 2020;55:2347–9.
Goedel P, Sieg N, Heger JM, Kutsch N, Herling C, Bärmann B-N, Scheid C, Borchmann
HU.Hematologic rescue of CAR-T cell mediated prolonged pancytopenia using autologous
peripheral blood hematopoietic stem cells in a lymphoma patient. Hema. 2021;5(3):e545.
Hill JA, Seo SK.How I prevent infections in patients receiving CD19-targeted chimeric antigen
receptor T cells for B-cell malignancies. Blood. 2020b;136(8):925–35.
Hill JH, Seo S.How I prevent infections in patients receiving CD19-targeted chimeric antigen
receptor T cells for B-cell malignancies. Blood. 2020a;136(8):925–35.
Jain T, etal. Hematopoietic recovery in patients receiving chimeric antigen receptor T-cell therapy
for hematologic malignancies. Blood Adv. 2020;4(15):3776–87.
Nastoupil LJ, Jain MD, Feng L, etal. Standard-of-care Axicabtagene Ciloleucel for relapsed or
refractory large B-cell lymphoma: results from the US lymphoma CAR-T consortium. J Clin
Oncol. 2020;38(27):3119–28.
Rejeski, K etal, CAR-HEMATOTOX: A model for CAR-T cell related hematological toxicity in
relapsed/refractory large B-cell lymphoma. Blood 2021a Jun 24; online ahead of print.
Rejeski K, Kunz WG, Rudelius M, Bücklein V, Blumenerg V, Schmidt C, Karschnia P, Schöberl F,
Dimitriadis K, Von Baumgarten L, Stemmler J, Weigert O, Dreyling M, Von Bergwelt-Baildon
M, Subklewe M.Severe Candida glabrata pancolitis and fatal Aspergillus fumigatus pulmo-
nary infection in the setting of bone marrow aplasia after CD19-directed CAR-T cell therapy- a
case report. BMC Infect Dis. 2021b;21(1):121.
Sterner R, Sakemura R, Cox M, Yang N, Khadka R, Forsman C, Hansen M, Jin F, Ayssou K, Hefazi
M, Schick K, Walters D, Chappell D, Ahmed O, Sahmoud t, Durrant C, Nevala W., Patnaik M,
Pease L, Hedin K, Kay N, Johnson A, Kenderian S.CM-SF inhibition reduces CRS and neuro-
inammation but enhances CAR-T cell function in xenografts. Blood. 2019;133(7):697–709.
Will Garner W, Samanta P, Haidar G. Invasive fungal infections after anti-CD19 chimeric anti-
gen receptor-modied T-cell therapy: state of the evidence and future directions. J Fungi.
2021;7:156.
Key Points
• Haematological toxicity is the most common adverse event after CD19-
specic CAR-T cell therapy and can predispose patients to severe infec-
tious complications.
• Administering prophylactic G-CSF from day 5 in neutropenic patients
does not increase the incidence of severe CRS/ICANS and is safe in pre-
serving CAR-T antilymphoma activity. Evidence level III-B.
• Post-CART immunosuppression is multifactorial (e.g., neutropenia, lym-
phopenia, steroid use, B-cell aplasia, and hypogammaglobulinaemia), and
infections signicantly contribute to non-relapse mortality.
• Anti-infective prophylaxis should follow institutional guidelines based on
the patient-specic risk factors.
M. Subklewe and R. Benjamin

155
Wudhikarn K, etal. DLBCL patients treated with CD19 CAR-T cells experience a high burden of
organ toxicities but low nonrelapse mortality. Blood Adv. 2020;4(13):3024–33.
Yakoub-Agha I, Chabannon C, Bader P, Basak GW, Bonig H, Ciceri F, Corbacioglu S, Duarte RF,
Einsele H, Hudecek M, Kersten MJ, Köhl U, Kuball J, Mielke S, Mohty M, Murray J, Nagler
A, Robinson S, Saccardi R, Sanchez-Guijo F, Snowden JA, Srour M, Styczynski J, Urbano-
Ispizua A, Hayden PJ, Kröger N. Management of adults and children undergoing chimeric
antigen receptor T-cell therapy: best practice recommendations of the European Society for
Blood and Marrow Transplantation (EBMT) and the joint accreditation committee of ISCT and
EBMT (JACIE). Haematologica. 2020;105(2):297–316.
Open Access
This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
29 Management ofMyelotoxicity (Aplasia) andInfectious Complications

157
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_30
H. Einsele
Department of Internal Medicine II, University Hospital Würzburg, Würzburg,
Bayern, Germany
e-mail: Einsele_h@ukw.de
I. Yakoub-Agha (
*)
Maladies du Sang, Unité de Thérapie Cellulaire, Centre hospitalier-Universitaire de Lille,
Lille, France
e-mail: [email protected]
30
Management ofOther Toxicities
HermannEinsele
andIbrahimYakoub-Agha
Secondary haemophagocytic lymphohistiocytosis (sHLH) or macrophage activa-
tion syndrome (MAS) is a life-threatening hyperinammatory syndrome that can
occur in patients with severe infections, e.g., COVID-19 infection, malignancy or
autoimmune diseases. It is also a rare complication of allogeneic haematopoietic
cell transplantation (allo-HCT), independent of the underlying trigger mechanism
or underlying disorders associated with high mortality. There have been increasing
reports of sHLH/MAS occurrence following CAR-T cell therapy, but its differentia-
tion from cytokine release syndrome (CRS) is often difcult (Sandler etal. 2020).
The diagnosis of sHLH/MAS post-HCT requires observation of the clinical
signs and symptoms of hyperinammation, which can overlap with the symptoms
of cytokine release syndrome or infectious complications, requiring a differential
diagnosis. Typically, these symptoms include fever, cytopenia of more than one
lineage, and multiorgan failure. Persistent fever without an identied infective cause
or worsening fever in patients who have been treated for infection should prompt
screening for sHLH/MAS (Karakike and Giamarellos-Bourboulis 2019). Serum
ferritin is a suitable and readily available biomarker of sHLH/MAS and can also be
used to monitor response to treatment.
CAR-T cell therapy, while emerging as an effective treatment for both haemato-
logical and nonhaematological malignancies, is associated with cytokine release
syndrome (CRS), an acute toxicity resulting in hyperinammation. Patients can
present with CRS across a spectrum of severities, from low-grade constitutional
symptoms to higher-grade systemic illness with multiorgan dysfunction, and in its

158
most severe form, CRS can progress to fulminant sHLH/MAS.Neelapu etal. pro-
posed diagnostic criteria for sHLH/MAS in patients with CRS post-CAR-T cell
therapy demonstrating a peak serum ferritin measurement of >10,000μg/L and two
of the following ndings: a grade>3 increase in serum transaminase or bilirubin;
grade > 3 oliguria or increase in serum creatinine; grade > 3 pulmonary oedema or
histological evidence of haemophagocytosis in the bone marrow or organs (Neelapu
etal. 2018) (Tables 30.1 and 30.2).
For effective treatment of sHLH/MAS, aggressive immunosuppression is
required to control the hyperinammatory state. Prompt recognition and treatment
are important and reduce mortality. Corticosteroids remain the cornerstone of induc-
tion treatment, although over half of patients are steroid resistant (Fukaya et al.
2008) (Table 30.3).
Anakinra, an IL-1 antagonist, is effective in refractory sHLH/MAS and relatively
safe in patients with sepsis (Shakoory et al. 2016) (Eloseily et al. 2019). Thus,
anakinra has been used for refractory sHLH/MAS and was found to be effective in
adult sHLH/MAS for patients with severe sHLH/MAS.Intravenous immunoglobu-
lin (IVIG) infusions may also be effective in steroid-resistant and infection (EBV)-
triggered sHLH/MAS (Chen etal. 1995).
A treatment protocol for sHLH/MAS accepting the heterogeneity of this syn-
drome has been recently published. The rst-line treatment is intravenous
Table 30.1 Use of published criteria to support the diagnosis of sHLH/MAS after CAR-T cell
therapy (adapted from Sandler etal. 2020)
Table 30.2 The Hscore adapted to CAR-T cell therapy
H. Einsele and I. Yakoub-Agha

159
methylprednisolone (IVMP) 1 g/day for 3–5 days plus IVIG 1 g/kg for 2 days,
which can be repeated on day 14. If there is evidence of established sHLH/MAS or
clinical deterioration, anakinra is added at 1–2mg/kg daily, increasing up to 8mg/
kg/day. CSA is considered for early or steroid-resistant disease. Etoposide should
be considered in refractory cases but can be problematic due to the already preexist-
ing cytopenias in patients with sHLH/MAS following CAR-T cell therapy.
Additionally, triggers, such as EBV, bacterial infection or underlying malignancy,
particularly lymphoma, should be screened for and treated if adequately dened
(Vatsayan etal. 2016).
Considerations forPatients Undergoing CAR-T Cell Therapy
• Steroids remain the cornerstone for sHLH/MAS treatment, but 50% of patients
are resistant.
• A recent recommendation for HLH/MAS after CAR-T therapy:
– Methylprednisolone 1g/day for 3–5days + IVIG 1g/kg for 2days, repeated
on day 14.
– In the case of deterioration, IV anakinra can be added up to 100mg x 4/day.
– Etoposide for refractory cases.
• Other treatments for sHLH/MAS after CAR-T cell therapy.
– Ruxolitinib: found to be effective in a case report and small phase 1 study
• Cytokine blockers might be used
• IVIG: might be effective, especially if the underlying cause is infection.
Table 30.3 Use of published protocols in the management of sHLH/MAS post-HSCT or CAR-T
cell therapy
Key Points
• Patients with persistent fever without an identied infection should be
screened for sHLH/MAS.
• Serum ferritin is a suitable and readily available biomarker of sHLH/MAS.
• Corticosteroids are the cornerstone of induction treatment.
• >50% of patients are steroid-refractory.
• IV anakinra is the second line treatment.
• Etoposide can be used in refractory sHLH/MAS.
30 Management ofOther Toxicities

160
References
Chen RL, Lin KH, Lin DT, Su IJ, Huang LM, Lee PI, etal. Immunomodulation treatment for child-
hood virus-associated haemophagocytic lymphohistiocytosis. Br J Haematol. 1995;89:282–90.
Eloseily EM, Weiser P, Crayne CB, Haines H, Mannion ML, Stoll ML, etal. Benet of anakinra
in treating pediatric secondary hemophagocytic lymphohistiocytosis. Arthritis Rheumatol.
2019;72:326–34.
Fukaya S, Yasuda S, Hashimoto T, Oku K, Kataoka H, Horita T, et al. Clinical features of
Haemophagocytic syndrome in patients with systemic autoimmune diseases: analysis of 30
cases. Rheumatology. 2008;47:1686–91.
Karakike E, Giamarellos-Bourboulis EJ.Macrophage activation-like syndrome: a distinct entity
leading to early death in sepsis. Front Immunol. 2019;10:55.
Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, et al. Chimeric anti-
gen receptor T-cell therapy– assessment and management of toxicities. Nat Rev Clin Oncol.
2018;15:47–62.
Sandler RD, Tattersall RS, Schoemans H, Greco R, Badoglio M, Labopin M, Alexander T,
Kirgizov K, Rovira M, Saif M, Saccardi R, Delgado J, Peric Z, Koenecke C, Penack O, Basak
G, Snowden JA.Diagnosis and Management of Secondary HLH/MAS Following HSCT and
CAR-T Cell Therapy in Adults; A Review of the Literature and a Survey of Practice Within
EBMT Centres on Behalf of the Autoimmune Diseases Working Party (ADWP) and Transplant
Complications Working Party (TCWP). Front Immunol. 2020;31(11):524.
Shakoory B, Carcillo JA, Chatham WW, Amdur RL, Zhao H, Dinarello CA, etal. Interleukin-1
receptor blockade is associated with reduced mortality in sepsis patients with features of
macrophage activation syndrome: reanalysis of a prior phase III trial. Critical Care Med.
2016;44:275–81.
Vatsayan A, Cabral L, Abu-Arja R.Hemophagocytic lymphohistiocytosis (HLH) after hematopoi-
etic stem cell transplant (HSCT) or an impostor: a word of caution! Blood Marrow Transplant.
2016;22:S262–3.
Open Access
This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
H. Einsele and I. Yakoub-Agha

161
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_31
U. Holtick (*)
Department I of Internal Medicine, University Hospital Cologne, Cologne, Germany
e-mail: [email protected]
E. Azoulay
Critical Care Department, Saint-Louis Hospital, Paris University, Paris, France
e-mail: [email protected]
31
ICU
UdoHoltick andElieAzoulay
CAR-T cell treatment comes with signicant side effects that challenge the struc-
ture and capacity of haematology wards and will regularly necessitate intermittent
patient transfer to the ICU.Life-threatening adverse events include cytokine release
syndrome and immune effector cell-associated neurotoxicity syndrome, which can
occur within hours or days after administration. Sepsis might also require ICU
admission within the days that follow CAR-T infusion in these high-risk immuno-
compromised patients.
Critical care and ICU specialists play an important role in the management of
patients receiving CAR-T therapies. A substantial number of patients require an
ICU bed, and CRS is the leading reason for ICU admission (Fitzgerald etal. 2017;
Gutierrez etal. 2020). Prompt and appropriate ICU management relies on a ne-
tuned dialogue between haematologists and ICU specialists and on an appropriate
denition of the threshold moment to target ICU admission. Hence, less than half of
patients require high-dose vasopressors, mechanical ventilation, or renal replace-
ment therapy (Azoulay etal. 2020). However, critical care also benets those in
whom appropriate antibiotics, source control of sepsis, echo-guided uid expan-
sion, prevention of acute kidney injury, and an optimal oxygenation strategy are
provided.
In some patients with comorbidities, the role of ICU specialists starts at the time
of patient eligibility for CAR-T cell therapy. Evaluation of patient frailty and risk
for developing organ dysfunction and sepsis helps dene the optimal timing of ICU
admission. When patients are starting lymphodepletion, the ICU specialists at least
receive a transmission. Of course, when patients have persistent stage 1 or stage 2

162
CRS, again, the ICU specialist is alerted. Overall, these careful strategies have
allowed a reduction in the need for ICU admission, with the numbers balanced with
widespread use of cell therapy and immunotherapy worldwide, which has been
helpful in a setting of scarce ICU beds.
To optimally manage CAR-T recipients, haematologists, oncologists, and inten-
sivists need to acquire the necessary knowledge and skills. Transdisciplinary meet-
ings ease harmonization of patient management, keeping all participants aware of
the advances in each specialty. Until recently, the ICU has primarily been used as a
bridge to cure patients with cancer (Azoulay etal. 2017; Gray etal. 2021). However,
CAR-T cell therapy challenges these concepts, producing a time-limited trial that is
offered to every CAR-T cell recipient, despite the underlying refractory malignancy,
and signicant hopes are put towards complete remission or bridging to another
promising therapy. However, not all patients respond to treatment with CAR-T
cells, and many patients ultimately relapse. Thus, we will need to adapt the approach
to admission and discharge from the ICU in a context of uncertainty and with hope
for recovery.
The key points below emphasize the role of the ICU specialist throughout the
CAR-T cell recipient journey and proposes the importance of maintaining tight col-
laboration across the involved specialties.
Key Points
CAR-T cell therapy: Framework to emphasize multidisciplinary
collaboration.
Adapted from Azoulay E etal., Am J Respir Crit Care Med. 2019
• Consultation with an ICU specialist to assess eligibility for CAR-T cell
therapy and anticipate post-infusion risks; consultation at the time of lym-
phodepletion and once any sign of toxicity or sepsis occurs.
• Apply a common information network to share important information.
• Reach agreement on the goals of care.
• Time-limited trials should be considered for all CAR-T cell recipients.
• CRS and ICANS must be assessed clinically several times per day for at
least 7days.
• Elicit prompt ICU admission once CRS reaches grade II.
• Leverage the latest advances.
• Liaise with all stakeholders to facilitate research.
• Share experiences with other specialists.
U. Holtick and E. Azoulay

163
References
Azoulay E, Darmon M, Valade S. Acute life-threatening toxicity from CAR-T cell therapy.
Intensive Care Med. 2020;46(9):1723–6. https://doi.org/10.1007/s00134- 020- 06193- 1.
Azoulay E, Schellongowski P, Darmon M, etal. The intensive care medicine research agenda
on critically ill oncology and hematology patients. Intensive Care Med. 2017;19 https://doi.
org/10.1007/s00134- 017- 4884- z.
Azoulay E, Shimabukuro-Vornhagen A, Darmon M, von Bergwelt-Baildon M. Critical Care
Management of Chimeric Antigen Receptor T Cell-related Toxicity. Be aware and prepared.
Am J Respir Crit Care Med. 2019;200(1):20–3. https://doi.org/10.1164/rccm.201810- 1945ED.
Fitzgerald JC, Weiss SL, Maude SL, et al. Cytokine release syndrome after chimeric antigen
receptor T cell therapy for acute lymphoblastic leukemia. Crit Care Med. 2017;45(2):e124–31.
https://doi.org/10.1097/CCM.0000000000002053.
Gray TF, Temel JS, El-Jawahri A.Illness and prognostic understanding in patients with hemato-
logic malignancies. Blood Rev. 2021;45:100692. https://doi.org/10.1016/j.blre.2020.100692.
Gutierrez C, Brown ART, Herr MM, etal. The chimeric antigen receptor-intensive care unit (CAR-
ICU) initiative: surveying intensive care unit practices in the management of CAR-T cell asso-
ciated toxicities. J Crit Care. 2020;58:58–64. https://doi.org/10.1016/j.jcrc.2020.04.008.
Open Access
This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
31 ICU

165
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_32
J. Gauthier (*)
Clinical Research Division, Fred Hutchinson Cancer Research Center, Seattle, WA, USA
Division of Medical Oncology, University of Washington, Seattle, WA, USA
e-mail: [email protected]
32
Post-CAR-T Cell Therapy (Consolidation
andRelapse): Acute Lymphoblastic
Leukaemia
JordanGauthier
Role of consolidative allogeneic haematopoietic cell transplantation (allo-HCT)
for B-cell acute lymphoblastic leukaemia (B-ALL) patients in minimal residual
disease- negative (MRD) complete remission (CR) after CD19 CAR-T cell
therapy.
The role of consolidative allo-HCT in B-ALL patients achieving MRD-negative
CR after CD19 CAR-T cell therapy is still debated. At the last update of the ELIANA
clinical trial, which investigated tisagenlecleucel in children and young adults with
relapsed or refractory (R/R) B-ALL, the median duration of remission and overall
survival was not reached (median follow-up, 24months). The 24-month relapse-
free survival probability in responders was 62%, with plateauing of the probability
curves after 1year (Grupp etal. 2019). Consolidation with allo-HCT was reported
in only 9% of CR patients, suggesting that CD19 CAR-T cell therapy alone with
tisagenlecleucel may be curative in a signicant proportion of paediatric patients. In
contrast, to date, the data do not suggest that CD19 CAR-T cell therapy is a deni-
tive approach in most adults with R/R B-ALL. Across the main academic and
industry- sponsored clinical trials of CD19 CAR-T cell therapy for adult B-ALL, the
median durations of response ranged from 8 to 19months, with important variations
in the proportion of patients receiving consolidative allo-HCT in CR after treatment
(35–75%) (Shah etal. 2019; Frey etal. 2020; Hay etal. 2019; Park etal. 2018). In
our experience, we observed favourable outcomes in patients undergoing allo-HCT
while in MRD-negative CR after dened-composition CD19 CAR-T cell therapy,
with 2-year EFS and OS probabilities of 61% and 72%, respectively. After adjusting
for previously identied prognostic factors for event-free survival (EFS; pre-
lymphodepletion LDH concentration and platelet count, cyclophosphamide-
udarabine lymphodepletion), the hazard ratio for allo-HCT was 0.39 (95% CI

166
0.13–1.15, p=0.09), suggesting a benecial effect on EFS.Based on these ndings,
our approach in adult patients is to recommend consolidative allo-HCT in adult
patients with R/R B-ALL in MRD-negative CR after CD19 CAR-T cell therapy.
Additionally, patient age and preferences, comorbidities, a history of prior trans-
plant, and MRD must be taken into account. Investigators from the NCI/NIH (Lee
etal. 2016) and Seattle Children’s Hospital (Summers etal. 2018) reported a sur-
vival advantage in children and young adults consolidated with allo-HCT after
CD19 CAR-T cell therapy. However, notably, to date, the available data rely on
nonrandomized, retrospective analyses, and are potentially subject to important
biases (Suissa 2007; Lévesque etal. 2010).
Management ofRelapsed B-ALL After CD19 CAR-T Cell Therapy
CD19-Positive Disease After CD19 CAR-T Cell Therapy
If cryopreserved end-manufacturing CAR-T cells are available and the target anti-
gen is still expressed, an attractive approach is to use the “left-over” cells to manu-
facture a second CAR-T cell product. We have shown that second CD19 CAR-T
cell infusions are feasible and well tolerated, but in most cases directed at the murine
single chain variable fragment (scFv) CAR domain, antitumour efcacy is limited
by anti-CAR immune responses. We observed superior outcomes in patients who
received cyclophosphamide and udarabine lymphodepletion prior to the rst
CAR-T cell infusion and who received a higher CAR-T cell dose (10 times higher
than the rst CAR-T cell infusion). However, second CAR-T cell infusions achieved
a CR in only 3 of 14 ALL patients (21%) (Gauthier etal. 2020). Efforts are under-
way to mitigate or circumvent anti-CAR immune responses using a CAR compris-
ing humanized or fully human scFvs (Gauthier etal. 2018; Brudno etal. 2020).
Maude etal. evaluated the use of the humanized scFv-bearing CD19 CAR-T cell
product CTL119in 38 children and young adults (Grupp etal. 2015). MRD-negative
CR could be achieved in murine CAR-exposed patients (43%), although at a lower
rate than in the CAR-naïve population (100%). The 12-month relapse-free survival
probabilities in responding patients were 82% and 56% in the CAR-exposed and
CAR-naïve cohorts, respectively. In another report, Cao etal. observed CR in 2 of 5
patients previously exposed to murine CD19 CAR-T cells(Cao etal. 2019). Further
studies are needed to determine whether immunogenicity, poor CAR-T cell func-
tion or disease-related factors underlie the reduced efcacy of CD19 CAR-T cells
employing fully human or humanized scFv in the murine CAR-exposed setting.
CD19-Negative Disease After CD19 CAR-T Cell Therapy
Encouraging results have been reported using CD22-targeted CAR-T cells, includ-
ing in patients with CD19-negative B-ALL blasts after prior CD19 CAR-T cell
therapy. Shah etal. reported their experience in 58 children and young adults with
J. Gauthier

167
R/R B-ALL, including 62% of patients who were previously treated with a CD19
CAR-T cell product (Shah et al. 2020). CD22 CAR-T cells achieved an MRD-
negative CR in 61% of cases, with a median duration of response of 6months (13
of 35 MRD-negative CR patients [37%] went on to receive allo-HCT). The MRD-
negative CR rate in patients previously treated with CD19 CAR-T cells was 64%. In
another report from Pan etal., CD22 CAR-T cells were administered to 34 children
and adult patients. Prior failure of CD19 CAR-T cell therapy was documented in
91% of patients. CD22 CAR-T cells achieved MRD-negative CRs in 76% of
patients. Responses were durable, with a 1-year leukaemia-free survival probability
of 58% in CR patients (11 of 30 CR patients [37%] went on to receive allo-HCT)
(Pan etal. 2019).
References
Brudno JN, Lam N, Vanasse D, Shen YW, Rose JJ, Rossi J, etal. Safety and feasibility of anti-
CD19 CAR-T cells with fully human binding domains in patients with B-cell lymphoma. Nat
Med. 2020;26(2):270–80.
Cao J, Cheng H, Qi K, Chen W, Shi M, Zheng J, etal. Humanized CD19-specic chimeric antigen
receptor T cells for acute lymphoblastic leukemia. Blood. 2019;134(Supplement_1):3872.
Frey NV, Shaw PA, Hexner EO, Pequignot E, Gill S, Luger SM, et al. Optimizing chimeric
antigen receptor T-cell therapy for adults with acute lymphoblastic leukemia. J Clin Oncol.
2020;38(5):415–22.
Gauthier J, Bezerra E, Hirayama AV, Pender BS, Vakil A, Steinmetz RN, etal. Repeat infusions
of CD19 CAR-T cells: factors associated with response, CAR-T cell invivo expansion, and
progression-free survival. ASTCT. 2020;26(3):S267–S8.
Gauthier J, Hirayama AV, Hay KA, Sheih A, Pender BS, Hawkins RM, etal. Immunotherapy with
T-cells engineered with a chimeric antigen receptor bearing a human CD19-binding single
chain variable fragment for relapsed or refractory acute lymphoblastic leukemia and B-cell
non-Hodgkin lymphoma. Blood. 2018;132(Supplement 1):1415.
Grupp SA, Maude SL, Rives S, Baruchel A, Boyer M, Bittencourt H, et al. Tisagenlecleucel
for the treatment of pediatric and young adult patients with relapsed/refractory acute lym-
phoblastic leukemia: updated analysis of the ELIANA clinical trial. Biol Blood Marrow Tr.
2019;25(3):S126–S7.
Key Points
• When feasible, allo-HCT should be offered to adult patients with R/R
B-ALL in MRD-negative CR after CD19 CAR-T cell therapy.
• CD22 CAR-T cells are associated with high response rates after CD19
CAR-T cell failure, and the most durable responses are observed after con-
solidative allo-HCT.
• CD19 CAR-T cells with fully human or humanized scFv are under inves-
tigation to mitigate anti-CAR immune responses, potentially impeding the
efcacy of repeat CAR-T cell infusions.
32 Post-CAR-T Cell Therapy (Consolidation and Relapse): Acute Lymphoblastic…

168
Grupp SA, Maude SL, Shaw PA, Aplenc R, Barrett DM. Durable remissions in children with
relapsed/refractory ALL treated with T cells engineered with a CD19-targeted chimeric antigen
receptor (CTL019). Blood. 2015;126(23):681.
Hay KA, Gauthier J, Hirayama AV, Voutsinas JM, Wu Q, Li D, et al. Factors associated with
durable EFS in adult B-cell ALL patients achieving MRD-negative CR after CD19 CAR-T cell
therapy. Blood. 2019;133(15):1652–63.
Lee DW, Stetler-Stevenson M, Yuan CM, Shah NN, Delbrook C, Yates B, etal. Long-term out-
comes following CD19 CAR-T cell therapy for B-ALL are superior in patients receiving a
Fludarabine/cyclophosphamide preparative regimen and post-CAR hematopoietic stem cell
transplantation. Blood. 2016;128(22):218.
Lévesque LE, Hanley JA, Kezouh A, Suissa S.Problem of immortal time bias in cohort studies:
example using statins for preventing progression of diabetes. BMJ. 2010;340(mar12 1):b5087.
Pan J, Niu Q, Deng B, Liu S, Wu T, Gao Z, et al. CD22 CAR-T cell therapy in refractory or
relapsed B acute lymphoblastic leukemia. Leukemia. 2019;33(12):2854–66.
Park JH, Riviere I, Gonen M, Wang X, Senechal B, Curran KJ, etal. Long-term follow-up of CD19
CAR-therapy in acute lymphoblastic leukemia. N Engl J Med. 2018;378(5):449–59.
Shah BD, Bishop M, Oluwole OO, Logan A, Baer MR, Donnellan W, etal. End of phase I results
of ZUMA-3, a phase 1/2 study of KTE-X19, anti-CD19 chimeric antigen receptor (CAR) T
cell therapy, in adult patients (pts) with relapsed/refractory (R/R) acute lymphoblastic leukemia
(ALL). ASCO. 2019;37(15_suppl):7006.
Shah NN, Highll SL, Shalabi H, Yates B, Jin J, Wolters PL, et al. CD4/CD8 T-cell selection
affects chimeric antigen receptor (CAR) T-cell potency and toxicity: updated results from a
phase I anti-CD22 CAR-T cell trial. J Clin Oncol. 2020;38(17):1938–50.
Suissa S. Immortal time bias in observational studies of drug effects. Pharmacoepidem Dr
S. 2007;16(3):241–9.
Summers C, Annesley C, Bleakley M, Dahlberg A, Jensen MC, Gardner R.Long term follow-up
after SCRI-CAR19v1 reveals late recurrences as well as a survival advantage to consolidation
with HCT after CAR-T cell induced remission. Blood. 2018;132(Supplement 1):967.
Open Access
This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
J. Gauthier

169
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_33
D. Blaise (*) · S. Fürst
Transplant and Cellular Immunotherapy Program, Department of Hematology, Institute
Paoli-Calmettes, Marseille, France
e-mail: [email protected].fr; [email protected]
33
Post-CAR-T Cell Therapy (Consolidation
andRelapse): Lymphoma
DidierBlaise andSabineFürst
Even after a decade of use, CAR-T cell therapy for non-Hodgkin lymphoma (NHL)
is still evolving, and disease control is now the main concern in the majority of
experienced centres. Indeed, despite highly appealing objective response (OR) rates
in refractory patients, the long-term overall survival (OS) of this population has
only slightly improved. Pivotal studies have suggested that 2-year OS rates do not
surpass 30%, even though results improve when complete response (CR) is achieved
within the rst 3months after treatment (Wang etal. 2020; Schuster etal. 2019;
Neelapu et al. 2017). Although achieving this exceptionally high level of OR is
praiseworthy, similar improvements have not been made regarding OS, and current
OS probabilities are not satisfactory. Of course, there are multiple reasons for this;
a substantial proportion of patients either do not achieve an initial response or expe-
rience progression very soon after treatment, with poor OS (Chow etal. 2019). Both
populations present with disease burden or aggressive cancer prior to CAR-T cell
therapy, possibly having been referred too late in the course of treatment or waited
too long before CAR-T cells were processed for them. Both of these issues have
potential solutions, such as more widely publicizing the efcacy of CAR-T cells,
which may increase referrals at an earlier stage, and developing methods, which are
already being heavily investigated, for shortening the manufacturing process (Raq
etal. 2020). In the latter case, the use of allogeneic lymphocytes could allow for
already prepared cells to be readily used when needed and would most likely be the
most efcient strategy as long as the risk of graft-versus host disease is offset
(Graham and Jozwik 2018). Thus, achieving CR is a crucial step in increasing OS,
as patients with partial response (PR) or stable disease (SD) present with lower OS,
while currently, recurrence appears to be rare when CR is maintained for more than
6months (Komanduri 2021). However, the disease will likely recur in more than

170
half of patients in the months following treatment, possibly due to issues such as the
poor persistence of CAR-T cells (which may not be as crucial as once thought for
acute lymphoblastic leukaemia (Komanduri 2021)) or the loss of target antigen
expression (which has been regularly documented (Raq etal. 2020)). Both of these
mechanisms could potentially be used to develop methods that reduce recurrence
after CAR-T cell therapy. In fact, the most popular approaches currently being
investigated are attempting to either use two CAR-T cell types that each target dif-
ferent antigens or to create CAR-T cell constructs that target either multiple anti-
gens or an antigen other than CD19 (Shah etal. 2020). The concomitant infusion of
CAR-T cells with targeted therapies is also being explored in other B-cell malignan-
cies and appears to both increase the CR rate and decrease recurrence (Gauthier
etal. 2020). When recurrence does occur, patient OS is rather dismal, and the best
remaining option would most likely be inclusion in a clinical trial. If this option is
not available, salvage therapy may be attempted, although cytotoxic treatments are
extremely limited given that most diseases have been refractory to numerous lines
of treatment prior to immunotherapy. A few case reports and studies with a small
patient population receiving anti-PD-1 antibodies, ibrutinib, or ImiDs have been
reported with largely anecdotal supporting evidence (Byrne etal. 2019). However,
even in the case of a new objective response (OR), the subsequent risk of recurrence
is substantial and may invite further consolidation with allogeneic haematopoietic
stem cell transplantation (Byrne etal. 2019), which has already been performed in
patients treated for acute lymphoblastic leukaemia (Hay etal. 2019). However, the
efcacy of this strategy remains to be validated in NHL patients in clinical trials.
Further supporting evidence, although limited, has recently been reported concern-
ing an additional treatment with CAR-T cells inducing an OR. Of the 21 NHL
patients included in the study, the OR rate after the second infusion was 52% (CR,
n = 4; PR, n = 7), with some durable responses inviting further investigations
(Gauthier etal. 2021). Overall, with such poor outcomes after recurrence, current
efforts are also focused on predicting the patients most likely to experience disease
progression and that are potential candidates for preemptive consolidation therapy,
although there is no doubt that patients who do not achieve a rapid CR should be the
rst candidates. Additionally, immune monitoring should encompass not only
CAR-T cell survival but also the detection of circulating tumour DNA (Komanduri
2021) because this could aid in detecting subclinical recurrence and in deciding
whether consolidation or maintenance therapy should be administered. However,
currently, all these approaches are highly speculative and require further clini-
cal study.
Key Points
• Relapse after CAR-T cell treatment for NHL is associated with a dismal
outcome.
• Presently, there is no consensus on salvage therapy.
• Investigating methods to prevent recurrence is mandatory.
• Inclusion in clinical trials is recommended.
D. Blaise and S. Fürst

171
References
Byrne M, Oluwole OO, Savani B, et al. Understanding and managing large B cell lymphoma
relapses after chimeric antigen receptor T cell therapy. Biol Blood Marrow Transplant.
2019;25(11):e344–51.
Chow VA, Gopal AK, Maloney DG, et al. Outcomes of patients with large B-cell lympho-
mas and progressive disease following CD19-specic CAR-T cell therapy. Am J Hematol.
2019;94(8):E209–13.
Gauthier J, Hirayama AV, Purushe J, etal. Feasibility and efcacy of CD19-targeted CAR-T cells
with concurrent ibrutinib for CLL after ibrutinib failure. Blood. 2020;135(19):1650–60.
Gauthier J, Bezerra ED, Hirayama AV, etal. Factors associated with outcomes after a second CD19-
targeted CAR-T cell infusion for refractory B-cell malignancies. Blood. 2021;137(3):323–35.
Graham C, Jozwik A. Pepper a et al allogeneic CAR-T cells: more than ease of access? Cell.
2018;7(10):5.
Hay KA, Gauthier J, Hirayama AV, et al. Factors associated with durable EFS in adult B-cell
ALL patients achieving MRD-negative CR after CD19 CAR-T cell therapy. Blood.
2019;133(15):1652–63.
Komanduri KV. Chimeric antigen receptor T-cell therapy in the management of relapsed non-
Hodgkin lymphoma. J Clin Oncol. 2021;39(5):476–86.
Neelapu SS, Locke FL, Bartlett NL, etal. Axicabtagene ciloleucel CAR-T cell therapy in refrac-
tory large B-cell lymphoma. N Engl J Med. 2017;377(26):2531–44.
Raq S, Hackett CS, Brentjens RJ.Engineering strategies to overcome the current roadblocks in
CAR-T cell therapy. Nat Rev Clin Oncol. 2020;17(3):147–67.
Schuster SJ, Bishop MR, Tam CS, etal. Tisagenlecleucel in adult relapsed or refractory diffuse
large B-cell lymphoma. N Engl J Med. 2019;380(1):45–56.
Shah NN, Johnson BD, Schneider D, et al. Bispecic anti-CD20, anti-CD19 CAR-T cells
for relapsed B cell malignancies: a phase 1 dose escalation and expansion trial. Nat Med.
2020;26(10):1569–75.
Wang M, Munoz J, Goy A, etal. KTE-X19 CAR-T cell therapy in relapsed or refractory mantle-
cell lymphoma. N Engl J Med. 2020;382(14):1331–42.
Open Access
This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
33 Post-CAR-T Cell Therapy (Consolidation andRelapse): Lymphoma

173
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_34
P. Rodríguez-Otero · J. F. SanMiguel (*)
Department of Hematology, Clínica Universidad de Navarra, University of Navarra,
Pamplona, Spain
e-mail: paurodriguez@unav.es; sanmiguel@unav.es
34
Post-CAR-T Cell Therapy (Consolidation
andRelapse): Multiple Myeloma
PaulaRodríguez-Otero andJesúsF.SanMiguel
Adoptive cell therapy with BCMA-directed autologous CAR-T cells has shown
very encouraging results in end-stage relapse and refractory multiple myeloma
(MM), with overall response rates ranging between 73% and 96.9%, complete
response (CR) rates between 33% and 67.9%, and MRD negativity in 50–74% of
patients in the two largest phase 2 studies of ide-cel (idecabtagene autoleucel,
KarMMa) and cilta-cel (ciltacabtagene autoleucel, CARTITUDE 1) reported thus
far (Madduri etal. 2020; Munshi etal. 2021). Unfortunately, responses are usually
not maintained, and no plateau has yet been seen in the survival curves. The median
progression-free survival (PFS) in the KarMMa study of ide-cel was 8.8months
(95% CI, 5.6–11.6) among all 128 patients infused, increased to 12.1months (95%
CI, 8.8–12.3) among patients receiving the highest dose (450×10
6
CAR+T cells)
and increased to 20.2months (95% CI, 12.3–NE) among those achieving a CR.In
the CARTITUDE-1 study, with a median follow-up of 12.4months, the median PFS
has not yet been reached, and the 12-month PFS rate was 76.6% (95% CI; 66.0–84.3).
The absence of a clear plateau in PFS differs from what has been observed in
DLBCL or B-ALL with currently approved CD-19-directed CAR-T cells, where
(albeit with a shorter PFS and lower rates of CR) patients remaining free from
relapse beyond 6 months are likely to enjoy prolonged disease control or even
be cured.
Mechanisms of resistance and relapse following CAR-T cell therapy in MM are
poorly understood, and several factors may explain these differences in survival
(D’Agostino and Raje 2020; Rodríguez-Otero etal. 2020). MM is a very heteroge-
neous disease with important clonal heterogeneity and a highly deregulated marrow
microenvironment. In addition, CAR-T cell therapy has been evaluated in very
heavily pretreated populations, with a signicant proportion of patients being

174
triple-class refractory and exposed to all available therapies, reecting a difcult-to-
treat population with an expected PFS of less than 4months (Gandhi etal. 2019).
To maintain responses and prolong survival, different strategies are being inves-
tigated, such as dual targeting to prevent antigen loss (Jiang etal. 2020) and manu-
facturing changes to increase the proportion of long-lived T cells with a memory
phenotype in the infused product, which has been associated with improved out-
comes and longer CAR-T cell persistence (Alsina etal. 2020; Costello etal. 2019;
Fraietta etal. 2018). The most advanced strategy to consolidate responses is combi-
nation with immunostimulatory drugs, such as IMIDs or checkpoint inhibitors, to
improve functional CAR-T cell persistence and avoid exhaustion. Several clinical
trials using these strategies are ongoing.
Interestingly, considering relapse after CAR-T cell therapy, emerging data show
a discordance between PFS and overall survival (OS). In the updated results from
the CRB-401 phase 1 study, the median PFS and OS reported were 8.8 (95% CI
5.9–11.9) and 34.2 (95% CI, 19.2–NE) months, respectively, across all doses (Lin
etal. 2020). OS data in the KarMMa study are still immature, with 66% of patients
censored overall (Munshi etal. 2021). Thus far, this gap between PFS and OS is not
as clear in other studies, but the follow-up time is still very short for the majority of
the trials. In the Legend-2 study, one of the BCMA studies with a longer follow-up
time, the median PFS for all treated patients was 20months, and the median OS was
not reached, with an 18-month OS rate of 68% (Chen etal. 2019). This suggests that
MM patients relapsing after CAR-T cell therapy may subsequently respond to sal-
vage treatments, including drug combinations that have previously failed. One can
speculate about potential modications of the immune system or the bone marrow
microenvironment induced by CAR-T cells. Unfortunately, data addressing this
phenomenon are not yet available. Furthermore, CAR-T cell therapy has been
shown to signicantly improve health-related quality of life (Cohen etal. 2020;
Martin III etal. 2020; Shah etal. 2020), and this better physical condition together
with a prolonged treatment-free interval are two key factors that may predispose
patients to accept additional rescue therapies, which would also contribute to the
OS gain.
Unfortunately, data are not yet available to elucidate what optimal rescue thera-
pies should be proposed after CAR-T cell progression. Anecdotal cases of patients
progressing after BCMA-directed CAR-T cell infusion and then treated with other
BCMA agents, such as belantamab mafodotin or checkpoint inhibitors, have been
reported and showed that this approach has limited efcacy (Cohen etal. 2019).
Nevertheless, the optimal approach in patients failing BCMA-directed CAR-T cell
therapy will be to employ therapies with different mechanisms of action (Melufen,
CELMODs, Selinexor) or immunotherapies directed against different targets, such
as SLAMF7, GPRC5D or FcRH5, using either bispecic T-cell engagers
(Talquetamab or Cevostamab) or even CAR-T cells. Indeed, new treatment modali-
ties and data from early phase studies including patients relapsing after CAR-T cell
therapy will provide the answer to this challenging problem: “Relapse following
BCMA CAR-T cell therapy: hope for further life.”
P. Rodríguez-Otero and J. F. SanMiguel

175
References
Alsina M, Shah N, Raje NS, etal. Updated results from the phase I CRB-402 study of anti-BCMA
CAR-T cell therapy bb21217in patients with relapsed and refractory multiple myeloma: cor-
relation of expansion and duration of response with T cell phenotypes. Blood. 2020;136(Suppl
1):25–6. https://doi.org/10.1182/blood- 2020- 140410.
Chen L, Xu J, Fu W Sr, etal. Updated phase 1 results of a rst-in-human open-label study of Lcar-
B38M, a structurally differentiated chimeric antigen receptor T (CAR-T) cell therapy targeting
B-cell maturation antigen (BCMA). Blood. 2019;134(Suppl_1):1858. https://doi.org/10.1182/
blood- 2019- 130008.
Cohen AD, Garfall AL, Dogan A, etal. Serial treatment of relapsed/refractory multiple myeloma
with different BCMA-targeting therapies. Blood Adv. 2019;3(16):2487–90. https://doi.
org/10.1182/bloodadvances.2019000466.
Cohen AD, Hari P, Htut M, et al. Patient expectations and perceptions of treatment in
CARTITUDE-1: phase 1b/2 study of ciltacabtagene autoleucel in relapsed/refractory multiple
myeloma. Blood. 2020;136(Suppl 1):13–5. https://doi.org/10.1182/blood- 2020- 136383.
Costello CL, Gregory TK, Ali SA, etal. Phase 2 study of the response and safety of P-BCMA-101
CAR-T cells in patients with relapsed/refractory (r/r) multiple myeloma (MM) (PRIME).
Blood. 2019;134(Suppl_1):3184. https://doi.org/10.1182/blood- 2019- 129562.
D’Agostino M, Raje N.Anti-BCMA CAR-T cell therapy in multiple myeloma: can we do better?
Leukemia. 2020;34(1):21–34. https://doi.org/10.1038/s41375- 019- 0669- 4.
Fraietta JA, Lacey SF, Orlando EJ, etal. Determinants of response and resistance to CD19 chi-
meric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med.
2018;24(5):563–71. https://doi.org/10.1038/s41591- 018- 0010- 1.
Gandhi UH, Cornell RF, Lakshman A, etal. Outcomes of patients with multiple myeloma refrac-
tory to CD38-targeted monoclonal antibody therapy. Leukemia. 2019;33(9):2266–75. https://
doi.org/10.1038/s41375- 019- 0435- 7.
Jiang H, Dong B, Gao L, etal. Clinical results of a multicenter study of the rst-in-human dual
BCMA and cd19 targeted novel platform fast CAR-T cell therapy for patients with relapsed/
refractory multiple myeloma. Blood. 2020;136(Suppl 1):25–6. https://doi.org/10.1182/
blood- 2020- 138614.
Key Points
• BCMA-directed CAR-T cell therapy shows very encouraging results in
triple- class refractory multiple myeloma populations, but there is not yet a
survival plateau.
• In the CRB-401 study, an important gap between PFS (median PFS of
8.8months) and OS (median OS of 34.2months) was observed, suggesting
that patients failing BCMA-directed CAR-T cell therapy may subsequently
respond to salvage treatments.
• Data are not yet available to elucidate what optimal rescue therapies should
be proposed after CAR-T cell progression.
• Salvage treatments after CAR-T cell treatment should include drugs with
new mechanisms of action (i.e., Melufen, Selinexor, CelMods) or target-
ing different antigens on the surface of plasma cells (i.e., GPRC5dD
(talquetamab) or FcRH5 (cevostamab).
34 Post-CAR-T Cell Therapy (Consolidation andRelapse): Multiple Myeloma

176
Lin Y, Raje NS, Berdeja JG, etal. Idecabtagene vicleucel (ide-cel, bb2121), a BCMA-directed
CAR-T cell therapy, in patients with relapsed and refractory multiple myeloma: updated
results from Phase 1 CRB-401 study. Blood. 2020;136(Suppl 1):26–7. https://doi.org/10.1182/
blood- 2020- 134324.
Madduri D, Berdeja JG, Usmani SZ, et al. CARTITUDE-1: phase 1b/2 study of ciltacabta-
gene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T cell ther-
apy, in relapsed/refractory multiple myeloma. Blood. 2020;136(Suppl 1):22–5. https://doi.
org/10.1182/blood- 2020- 136307.
Martin T III, Lin Y, Agha M, etal. Health-related quality of life in the cartitude-1 study of cilt-
acabtagene autoleucel for relapsed/refractory multiple myeloma. Blood. 2020;136(Suppl
1):41–2. https://doi.org/10.1182/blood- 2020- 136368.
Munshi NC, Anderson LDJ, Shah N, etal. Idecabtagene vicleucel in relapsed and refractory mul-
tiple myeloma. N Engl J Med. 2021;384(8):705–16. https://doi.org/10.1056/NEJMoa2024850.
Rodríguez-Otero P, Prósper F, Alfonso A, etal. CAR-T cells in multiple myeloma are ready for
prime time. J Clin Med. 2020;9(11) https://doi.org/10.3390/jcm9113577.
Shah N, Delforge M, San-Miguel JF, et al. Secondary quality-of-life domains in patients with
relapsed and refractory multiple myeloma treated with the BCMA-directed CAR-T cell ther-
apy idecabtagene vicleucel (ide-cel; bb2121): results from the Karmma clinical trial. Blood.
2020;136(Suppl 1):28–9. https://doi.org/10.1182/blood- 2020- 136665.
Open Access
This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
P. Rodríguez-Otero and J. F. SanMiguel

177
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_35
S. C. Berger (*) · B. Fehse
Department of Stem Cell Transplantation (SCT) and Research Department Cell and Gene
Therapy, University Medical Center Hamburg-Eppendorf (UKE), Hamburg, Germany
e-mail: [email protected]; fehse@uke.de
M.-T. Rubio
Service d’Hematolgie, Hôpital Brabois, CHRU Nancy and Biopole de l’Université de
Lorraine, Vandoeuvre les Nancy, France
e-mail: m.rubio@chru-nancy.fr
35
Immune Monitoring
SusannaCarolinaBerger, BorisFehse,
andMarie-ThérèseRubio
CAR-T cell expansion and persistence are critical parameters for therapeutic ef-
cacy and toxicity (Locke etal. 2020). However, CAR-T cells are patient-specic
‘living drugs’ with an unpredictable ability to expand invivo. Thus, close postinfu-
sion monitoring should be a major prerequisite to better manage this therapy.
Critical parameters include CAR-T cell expansion kinetics and phenotype immune
reconstitution and serum biomarkers (Fig.35.1; Kalos etal. 2011; Hu and Huang
2020). Additionally, prospective collection and storage of patient specimens should
be planned for future hypothesis-driven studies at specialized research centres. To
date, despite the rapid expansion of CAR-T cell therapy, no standard recommenda-
tions exist for CAR monitoring, and harmonization of efforts across multiple cen-
tres is urgently needed.
Molecular Monitoring ofCAR-T Cells via Digital PCR (dPCR)
Most clinically used CAR-T cell products consist of autologous lymphocytes stably
transduced with retro- or lentiviral vectors encoding the respective CAR construct.
Integrated CAR vectors are commonly detected at the genomic level using real-time
quantitative PCR (qPCR) or dPCR.Surprisingly, outside of clinical trials, CAR-
specic diagnostic tools were initially missing, requiring the de novo design of lab-
made specic assays to enumerate CAR-T cells invivo (Badbaran etal. 2020; Fehse

178
etal. 2020; Kunz etal. 2020). Despite technological differences, both qPCR and
dPCR assays yield robust and accurate results, with limited requirements regarding
sample quality (Table35.1).
dPCR is extremely sensitive and does not rely on standard curves or multiple
repetitions. As a limitation, DNA-directed PCR monitoring provides no information
on the expression of the CAR construct. However, the expansion of CAR-T cells
strongly depends on the interaction of the CAR with its cognate antigen. In accor-
dance, our data have shown excellent correlation of dPCR with ow cytometry
(Badbaran etal. 2020) as well as clinical (Ayuk etal. 2021) results. Because ow
cytometry-based assays facilitate phenotypic characterization of CAR-T cells, the
two methods complement each other well.
Flow Cytometry Monitoring ofCAR-T Cells
Identication of CAR-T cells by ow cytometry (CMF) can be performed by using
monoclonal antibodies (mAbs) directly recognizing the CAR (idiotype, linker
region) or a specic tag included in the CAR construct. Alternatively, indirect detec-
tion can be achieved using antigen-Fc chimeric proteins containing the CAR target
antigen fused to a human IgG Fc fragment. A secondary staining step is required for
the detection of CAR-expressing cells with an anti-Fc or anti-biotin (if the antigen-
Fc is biotinylated) mAb labelled with a uorochrome (Hu and Huang 2020). In
practice, outside of clinical trials, patients receiving commercial CD19 CAR-T cells
are monitored with biotinylated CD19-Fc proteins in a two-step staining protocol.
The advantage of CMF is the possibility of combining CAR staining with other cell
surface markers to characterize CAR-T cells in terms of T cell subtype (CD4 and
CD8 expression), differentiation (naïve versus memory), and exhaustion (PD1,
TIM3, Lag3). In addition, the results can be provided in real time to physicians. The
limitation of the technique is the relatively low sensitivity. Below 0.5% of T cells,
the reliability of CMF is weak and justies pursuing monitoring via PCR. Two
important pieces of information can be obtained with sequential CMF analysis of
CAR-T
cells
Cy/Flu
Disease Assessment
Da
y0 35 100 180 365**
‘Diagnostic’ Monitoring:
‘Research’ Monitoring:
CAR-T-cell expansion kinetics and phenotype
Immune reconstitution
Functional Analysis
Transgene-product specific immunity
Next-generation sequencing (e.g. TCR, RNASeq)
Multiplex immunfluorescent histopathology
*BM and/or Tumor
** Follow-up up to 15 year
s
*
Fig. 35.1 Schematic overview of monitoring after CAR-T cell therapy
S. C. Berger et al.

179
CAR-T cells in the peripheral blood after cell infusion: the expansion peak (Cmax,
maximum CAR-T cell rate in percentage or absolute value) and the area under the
curve of circulating CAR-T cells between D0 and D28 (AUC0-28). These two
parameters have been associated with the response and the risk of complications
after treatment in B-lymphoid malignancies (Park etal. 2018; Fraietta etal. 2018;
Locke etal. 2020; Ayuk etal. 2021). To determine these parameters, the recom-
mended frequency of CAR-T cell monitoring is two or three times a week for the
rst 2weeks after CAR-T cell administration, on days 21 and 28, once a month until
3months and then every 3months until 1year (Rubio etal. 2021).
Monitoring ofAdditional Immune Parameters (Non-CAR-T, B,
andNK Cells andCytokines)
Patients receiving anti-CD19 CAR-T cells might develop prolonged T CD4 lym-
phopenia as well as B-cell aplasia with severe hypogammaglobulinaemia, making
them particularly susceptible to bacterial and viral infections even after
Table 35.1 Comparison of molecular monitoring tools
qPCR
dPCR
Principle of analysis
Target-specic primer and
uorescent probes
Yes Yes
Analysis of the gene of
interest within the sample
At the population level After partitioning into tiny
droplets
Amplication Amplies and quanties
amplicon over PCR cycles
Amplies and quanties
amplicon separately within a
droplet
Quantication Continuous intermediate
uorescence measurements
Relies on end-point
uorescence
Data robustness and
reliability
High Very High
a
Requirement of a reference
sample/standard curve
Yes No
Single cell tools No Yes
Overall properties
Distribution of the
instruments
Commonly available Still less frequently available
Handling Easy to implement Requires more training &
technical skills
Cost (Campomenosi etal.
2016)
b
3.33 € per sample 3.66 € per sample
Multiplexing
c
Yes Yes
Certied instruments
commercially available?
Yes Yes (limited)
a
Compared to qPCR, very robust amplication kinetics and suppresses amplication noise
b
In the cited study, costs per sample were based on single measurements for dPCR vs. triplicate
analyses for qPCR.They did not include instrument amortization
c
Decreases sample/reagent use and pipetting noise, increases throughput
35 Immune Monitoring

180
haematopoietic recovery (Logue etal. 2021). Therefore, routine immune surveil-
lance of non-CAR-T CD4 and CD8 T cells, B cells, and NK cells and the levels of
serum immunoglobulins is recommended during the rst year of follow-up.
Many cytokines are produced in large quantities after CAR-T cell administration
as a result of activation of T lymphocytes (IL-6, IFN-γ, sIL2-Rα, sIL-6R, GM-CSF,
IL-2, and TNF-α), activation and attraction of mono-macrophages (IFNα, IL-1β,
IL-6, IL1Rα, IL10, IL-12, IL-13, IL-15, sIL6-R, TNF-α, CXCL10, CCL2, and IL-8)
and in response to tissue damage (IL-6, IL-8, G-CSF, and GM-CSF) (Brudno and
Kochenderfer 2019). Confounding factors, such as sepsis, degree of CAR-T cell
expansion and tumour burden, also impact cytokine levels. Some cytokine signa-
tures have been described to predict the occurrence of cytokine release syndrome
(CRS) (Teachey et al. 2016), immune effector cell-associated neurotoxicity syn-
drome (ICANS) (Santomasso etal. 2018) or the expansion capacity of CAR-T cells
invivo (Kochenderfer etal. 2017). One major limitation in clinical practice is the
absence of a validated fast cytokine quantication test predicting severe complica-
tions. Therefore, further studies are required in homogeneous groups of patients to
determine whether cytokines can predict the occurrence of complications or treat-
ment efcacy. Participation in prospective studies or collection of serum at each
time point of CAR-T cell analysis is recommended.
Key Points
Immune monitoring after CAR-T cell therapy should be carefully performed:
• Medical CAR-T cell products are complex ‘living drugs’ with unpredict-
able invivo performance. Thus, the establishment of accompanying diag-
nostic and research monitoring programmes is a priority for rational
development of this approach.
• Molecular monitoring, especially dPCR, is an excellent, robust, and sensi-
tive tool for real-time/on-site persistence tracking.
• Flow cytometry is an easy and rapid tool to monitor early CAR-T cell
expansion and characterize CAR-T cell phenotype, both of which have
been correlated with the response.
• Routine monitoring of T, B, and NK cell populations and immunoglobulin
levels is recommended to evaluate infection risk.
• Serum collection is recommended to further explore and identify cytokine
signatures that enable prediction of complications or response.
• Efforts to harmonize patient monitoring across multiple centres following
CAR-T cell infusion would be desirable (i.e., reference labs, shared data-
bases, and collaborations with dedicated centres for ‘next generation’
research). Successful implementation of these joint efforts will greatly
advance our understanding of the biology involved in transferring CAR-T
cells and, most importantly, serve our patients.
S. C. Berger et al.

181
References
Ayuk FA, Berger SC, Badbaran A, etal. Axicabtagene ciloleucel invivo expansion and treatment
outcome in aggressive B-cell lymphoma in a real-world setting. Blood Adv. 2021;5(11):2523–7.
Badbaran A, Berger C, Riecken K, etal. Accurate in-vivo quantication of CD19 CAR-T cells
after treatment with axicabtagene ciloleucel (Axi-cel) and tisagenlecleucel (Tisa-cel) using
digital PCR.Cancers (Basel). 2020;12:1970.
Brudno JN, Kochenderfer JN.Recent advances in CAR-T cell toxicity: mechanisms, manifesta-
tions and management. Blood Rev. 2019;34:45–55.
Campomenosi P, Gini E, Noonan DM, etal. A comparison between quantitative PCR and droplet
digital PCR technologies for circulating microRNA quantication in human lung cancer. BMC
Biotechnol. 2016;16:60.
Fehse B, Badbaran A, Berger C, et al. Digital PCR assays for precise quantication of CD19-
CAR- T cells after treatment with axicabtagene ciloleucel. Mol Ther Methods Clin Dev.
2020;16:172–8.
Fraietta JA, Lacey SF, Orlando EJ, etal. Determinants of response and resistance to CD19 chi-
meric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med.
2018;24:563–71.
Hu Y, Huang J.The chimeric antigen receptor detection toolkit. Front Immunol. 2020;11:1770.
Kalos M, Levine BL, Porter DL, etal. T cells with chimeric antigen receptors have potent anti-
tumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med.
2011;3:95ra73.
Kochenderfer JN, Somerville RPT, Lu T, etal. Lymphoma remissions caused by anti-CD19 chime-
ric antigen receptor T cells are associated with high serum interleukin-15 levels. J Clin Oncol.
2017;35:1803–13.
Kunz A, Gern U, Schmitt A, etal. Optimized assessment of qPCR-based vector copy numbers as
a safety parameter for GMP-grade CAR-T cells and monitoring of frequency in patients. Mol
Ther Methods Clin Dev. 2020;17:448–54.
Locke FL, Rossi JM, Neelapu SS, et al. Tumor burden, inammation, and product attributes
determine outcomes of axicabtagene ciloleucel in large B-cell lymphoma. Blood Adv.
2020;4:4898–911.
Logue JM, Zucchetti E, Bachmeier CA, et al. Immune reconstitution and associated infec-
tions following axicabtagene ciloleucel in relapsed or refractory large B-cell lymphoma.
Haematologica. 2021;106(4):978–86.
Park JH, Riviere I, Gonen M, etal. Long-term follow-up of CD19 CAR therapy in acute lympho-
blastic leukemia. N Engl J Med. 2018;378:449–59.
Rubio MT, Varlet P, Allain V, et al. Immunomonitoring of patients treated with CAR-T cells
for hematological malignancy: guidelines from the CARTi group and the francophone
Society of Bone Marrow Transplantation and Cellular Therapy (SFGM-TC). Bull Cancer.
2021;108(12S):S53–64.
Santomasso BD, Park JH, Salloum D, etal. Clinical and biological correlates of neurotoxicity
associated with CAR-T cell therapy in patients with B-cell acute lymphoblastic leukemia.
Cancer Discov. 2018;8:958–71.
Teachey DT, Lacey SF, Shaw PA, etal. Identication of predictive biomarkers for cytokine release
syndrome after chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia.
Cancer Discov. 2016;6:664–79.
35 Immune Monitoring

182
Open Access
This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
S. C. Berger et al.

183
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_36
P. Hayden (*)
Department of Haematology, Trinity College Dublin, St. James’s Hospital, Dublin, Ireland
e-mail: [email protected]
N. Gagelmann
Department of Stem Cell Transplantation, University Medical Center Hamburg,
Hamburg, Germany
e-mail: [email protected]
J. Snowden
Department of Haematology, Shefeld Teaching Hospitals NHS Foundation Trust,
Shefeld, UK
Department of Oncology and Metabolism, The University of Shefeld, Shefeld, UK
e-mail: [email protected]
36
Long-Term Follow-Up andLate Effects
PatrickHayden, NicoGagelmann, andJohnSnowden
Little is known about the long-term effects of CAR-T cell therapy. Although
medium-term complications, such as cytopenia and hypogammaglobulinaemia,
may persist and require ongoing treatment, there do not appear to be other durable
toxicities specic to this new immunotherapeutic class (Fried etal. 2019; Cordeiro
etal. 2020; Cappell etal. 2020). However, to date, CAR-T therapy has been evalu-
ated in patients with multiple relapsed diseases following several lines of treatment,
including allogeneic stem cell transplantation, making it difcult to identify which
effects may be directly attributable to this novel treatment. Nonetheless, as the use
of CAR-T cell therapy increases, structured models for survivorship care will need
to be established. The factors that will affect care requirements include the primary
malignancy, prior treatment, the specic CAR-T therapy and patient age and frailty.
The main late effects identied to date are shown in Table 36.1.
Hypogammaglobulinaemia and prolonged cytopenias appear to be more common in
patients with ALL than in patients with B-NHL.In the ELIANA trial, which tested
tisagenlecleucel (Kymriah™) in ALL, the median time to B-cell recovery was not
reached at a median follow-up time of 13months (Maude etal. 2018). Prolonged
cytopenias in all three cell lines have also been commonly reported. In an Israeli

184
study of 29 patients with either ALL or B-NHL responding after treatment with
CTL109 with a CD28 costimulatory domain, factors associated with late cytopenias
were prior allo-HCT and higher-grade CRS (Fried etal. 2019).
Apart from one patient in the ZUMA-1 trial who developed MDS at 19months,
there were no secondary malignancies reported in the three clinical trials that led to
licensing of CD-19-directed therapy in B-ALL and B-NHL.In addition, some late
cancers are to be expected in such heavily pretreated patients. Although there is one
report of unintended insertion of the CAR gene into leukaemic B cells, thus far,
there have been no reports of insertional oncogenesis during CAR-T cell production.
The role of vaccinations following CAR-T cell therapy remains unclear. Until
evidence-based specic CAR-T vaccination programmes are produced, protocols
similar to HSCT should be considered (Majhail etal. 2012).
Follow-Up andProgrammes
As a specialized service, CAR-T therapy in Europe is generally provided based on
a hub-and-spoke model: patients are referred from local hospitals to regional cellu-
lar therapy centres. One option is to provide follow-up in JACIE-accredited allo-
HCT late effects clinics alongside transplant recipients. They operate on a checklist
model to ensure that survivors are systematically and longitudinally assessed for
late toxicities. Over time, dedicated CAR-T late effects clinics can be developed if
the growing pool of survivors reaches a critical mass. Service-level agreements
(SLAs) between CAR-T centres and referral centres should cover shared care and
outreach arrangements.
Table 36.1 Main late effects after CAR-T cell therapy
Effect
Occurrence
Management
Cytopenia (esp. neutropenia)
All grades
>2
Two months after infusion:
~50%
~20%
Transfusion, growth factors,
infection prophylaxis
Hypogammaglobulinaemia ~50%, prolonged years after
infusion
Intravenous
immunoglobulins (IVIGs)
Infections Predominantly upper
respiratory tract, >50%
bacterial
IVIGs, antibiotics, viral
screening, vaccination
Secondary malignancies Solid tumour>haematological Surveillance, awareness
Neurological effects ~10%, neuropathy and
cerebrovascular events
Supportive care,
interdisciplinary approach to
diagnosis and therapy
Psychiatric issues ~10%, depression and anxiety
Immune-related issues <10%, alveolitis, pneumonitis,
dermatitis, arthralgia, and
myositis, etc.
Corticosteroids,
immunosuppression,
interdisciplinary approach to
diagnosis and therapy
P. Hayden et al.

185
Such clinics require multidisciplinary team (MDT) input, including physicians
involved in CAR-T administration, clinical nurse specialists, clinical psychologists,
data managers, and clinical trial staff. All CAR-T recipients will have been heavily
pretreated. Therefore, a cumulative burden of broader physical and psychological
late effects will need to be considered. Areas to cover in the clinic include CAR-T
persistence; secondary malignancies; autoimmune disease; endocrine, reproductive
and bone health; psychological health; and patient-reported outcomes, including
quality of life (Buitrago etal. 2019; Ruark etal. 2020). Importantly, the patient-
reported quality-of-life studies performed thus far indicate levels of physical and
mental health comparable to that in the normal population.
Initial follow-up will be determined by the status of the underlying disease.
Patients should be seen monthly for the rst year, when the focus will be on remis-
sion status alongside any short-term complications. Subsequent follow-up can focus
on longer-term effects, 6 months for the following 2 years, annually until the
15 year, and potentially indenitely. Patients who proceed to subsequent HSCT,
cytotoxic therapy and/or immune effector cell therapy should be followed as recom-
mended by Majhail etal. (2012).
Post-authorisation Safety Surveillance (PASS)
As both tisagenlecleucel (Kymriah™) and axicabtagene ciloleucel (Yescarta™) are
based on genetic modication of autologous T-cells using viral vectors, the EMA
and FDA made marketing approval conditional on a 15-year PASS.In 2019, the
cellular therapy module of the EBMT registry was found by the EMA to be t-for-
purpose for regulatory oversight of such pharmacoepidemiological studies. The
MED-A cell therapy form has been modied for use with CAR-T cells and other
academic- or industry-manufactured cell therapies. In November 2020, the German
health insurance regulator directed centres to report commercial CAR-T cell treat-
ments to the EBMT Registry and conrmed that reporting such data will be a condi-
tion for reimbursement of the costs of CAR-T cell therapy.
JACIE
FACT-JACIE standards were initially developed for the accreditation of HCT pro-
grammes (Snowden etal. 2017; Saccardi etal. 2019). The current seventh edition of
the standards also covers immune effector cells (IECs) to accommodate cellular
therapy, including CAR-T cells. In addition to clauses addressing the need for poli-
cies on the management of acute toxicities, standard B.7.12 species the need for
“policies and Standard Operating Procedures for monitoring by appropriate special-
ists of recipients for post-cellular therapy late effects”. Inspection of IEC standards
is incorporated within standard JACIE site visits.
For centres that undertake CAR-T cell therapy outside of an accredited allo-HCT
programme, there are a number of options. Given that most CAR-T cell therapies
36 Long-Term Follow-Up andLate Eects

186
are currently used to treat lymphoma, compliance with the IEC standards can be
achieved as part of the accreditation covering autologous HCT (auto-HCT). The
same considerations could apply to myeloma specialists working outside of allo-
HCT programmes. In the event of CAR-T cells or related therapies becoming appli-
cable more broadly to nonhaematological cancers, an alternative strategy already
adopted by FACT is to undertake independent IEC accreditation specically for
CAR-T cells and other IECs. JACIE also provides a robust method to ensure that
programmes meet the requirements for mandatory long-term data submission to the
EBMT Registry, as well as potential benchmarking of survival outcomes.
The eighth edition of the FACT-JACIE standards will be published in 2021 with
more detail on immune effector cells to help provide a framework for centres to
establish and assure the quality and safe practice of treatment administration and
short- and long-term follow-up of CAR-T therapy.
References
Buitrago J, Adkins S, Hawkins M, Iyamu K, Oort T.Adult survivorship: considerations following
CAR-T cell therapy. Clin J Oncol Nurs. 2019;23(2):42–8.
Cappell KM, Sherry RM, Yang JC, Goff SL, Vanasse DA, McIntyre L, etal. Long-term follow-
up of anti-cd19 chimeric antigen receptor T-cell therapy. J Clin Oncol. 2020;38(32):3805–15.
Cordeiro A, Bezerra ED, Hirayama AV, Hill JA, Wu QV, Voutsinas J, etal. Late events after treat-
ment with CD19-targeted chimeric antigen receptor modied T cells. Biol Blood Marrow
Transplant. 2020;26(1):26–33.
Fried S, Avigdor A, Bielorai B, Meir A, Besser MJ, Schachter J, etal. Early and late hematologic
toxicity following CD19 CAR-T cells. Bone Marrow Transplant. 2019;54(10):1643–50.
Key Points
• To date, few durable toxicities have been directly attributable to CAR-T
cell therapy.
• The principal late effects identied to date include hypogammaglobulinae-
mia, cytopenias, and infections.
• Structured models for survivorship care include JACIE-accredited allo-
HCT late effects clinics.
• Areas to monitor in the clinic include CAR-T persistence; secondary
malignancies; autoimmune disease; endocrine, reproductive and bone
health; psychological health; and patient-reported outcomes, including
quality of life.
• EMA has mandated 15-year postauthorization safety surveillance (PASS)
of all CAR-T cell therapies, and the cellular therapy module of the EBMT
registry has been approved for this purpose.
• The current seventh edition of the FACT-JACIE transplant standards also
covers immune effector cells (IECs) to accommodate cellular therapy,
including CAR-T cells.
P. Hayden et al.

187
Majhail NS, Rizzo JD, Lee SJ, Aljurf M, Atsuta Y, Bonm C, etal. Recommended screening
and preventive practices for long-term survivors after hematopoietic cell transplantation. Bone
Marrow Transplant. 2012;47(3):337–41.
Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, etal. Tisagenlecleucel in chil-
dren and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439–48.
Ruark J, Mullane E, Cleary N, Cordeiro A, Bezerra ED, Wu V, etal. Patient-reported neuropsychi-
atric outcomes of long-term survivors after chimeric antigen receptor T cell therapy. Biol Blood
Marrow Transplant. 2020;26(1):34–43.
Saccardi R, McGrath E, Snowden AJ.JACIE accreditation of HSCT programs. In: The EBMT
handbook; 2019. p.35–40.
Snowden JA, McGrath E, Duarte RF, Saccardi R, Orchard K, Worel N, etal. JACIE accreditation
for blood and marrow transplantation: past, present and future directions of an international
model for healthcare quality improvement. Bone Marrow Transplant. 2017;52(10):1367–71.
Open Access
This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
36 Long-Term Follow-Up andLate Eects

Part V
Access to CAR-T Cells

191
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_37
E. McGrath
EBMT, Barcelona, Spain
e-mail: [email protected]
P. Machalik (
*)
Be The Match BioTherapies, Minneapolis, MN, USA
e-mail: [email protected]
37
The Regulatory Framework forCAR-T
Cells inEurope: Current Status
andForeseeable Changes ANDCentre
Qualification by Competent Authorities
andManufacturers
EoinMcGrath andPetrMachalik
Current Framework
Under current European Union regulations, CAR-T cell therapies fall under the
advanced therapy medicinal products (ATMPs) framework. ATMPs represent a cat-
egory of medicinal products dened in EU Regulation 1394/2007 and subdivided
into four categories, of which autologous or allogeneic CAR-T cells, among other
therapies, are considered gene therapy medicinal products (GTMPs). ATMPs are
subject to a centralized evaluation framework whereby one authorization is valid for
all countries in the EU led by the European Medicines Agency’s Committee for
Advanced Therapies (CAT). The framework includes different regulatory pathways
for bringing ATMPs from clinical trials to market authorization, and the regulatory
pathway taken will depend on a product’s characteristics and the target patient pop-
ulation. In 2018, two chimeric antigen receptor (CAR) T cell therapies, Yescarta and
Kymriah, completed their authorization process via the priority medicines PRIME
scheme to Marketing Authorization (Detela and Lodge 2019).
The production, distribution, and administration of ATMPs require a completely
different organization plan than that used for HSCT, with manufacturing typically at
a central facility in compliance with good manufacturing practices (GMPs), a ver-
sion of which was released in 2017 by the European Commission to specically

192
deal with the manufacturing of ATMPs.
1
Since a majority of the ATMPs that prog-
ress to authorization or at least to clinical trials are manufactured from autologous
mononuclear cells, starting material is usually procured by hospital- or blood bank-
operated apheresis facilities, creating a peculiar situation in which a product starts
under one regulation—EU Tissues and Cells Directives
2
—before passing to
another—ATMP Regulation- and in which a hospital acts as a service provider to
industry, an interaction that requires further denition of the respective responsibili-
ties and liabilities (McGrath and Chabannon 2018). The Tissues and Cells Directives,
which cover all steps in the transplant process from donation, procurement, testing,
processing, preservation, storage, and distribution, are undergoing a review that is
expected to lead to a legislative proposal by the European Commission in late 2021,
and it is anticipated that the new framework will further consider how products
cross the interface between the two frameworks.
Given the high toxicity prole of CAR-T cell therapies, marketing authorization
may be subject to conditions that lead to a risk management plan (RMP). The RMP
for the currently authorized CAR-T therapies includes the need for manufacturers to
qualify the sites that will treat patients. Site qualication is addressed below.
Hospital Exemption
In recognizing that many potential ATMPs are used for limited numbers of patients
and with little commercial interest, Regulation 1394/2007 created the so-called hos-
pital exemption (HE) under Article 28, exempting from authorization requirements
those ATMPs manufactured in hospitals or universities where the medicine is pre-
scribed for individual patients under the care of a medical practitioner. This manu-
facturing should occur on a nonroutine basis according to specic quality standards
(GMPs) and only within the same member state.
In February 2021, the Spanish pharmaceutical regulator AEMPS authorized the
rst CAR-T cell therapy approved by a European national authority under the hos-
pital exemption clause for the ARI-0001 CAR-T developed by the Hospital Clinic
in Barcelona.
3
National authorities oversee the approval of HE products, which has resulted in
signicant variations between member states in how approval is applied, in turn
leading to criticism from both industry and academia that the approval process is
unclear and inconsistent.
1
Guidelines of 22.11.2017 Good Manufacturing Practice for Advanced Therapy Medicinal
Products. EudraLex The Rules Governing Medicinal Products in the European Union Volume 4
Good Manufacturing Practice.
2
Directive 2004/23 of the European Parliament and of the Council of 31 March 2004 on setting
standards of quality and safety for the donation, procurement, testing, processing, preservation,
storage, and distribution of human tissues and cells.
3
https://www.aemps.gob.es/informa/notasinformativas/medicamentosusohumano-3/2021-
medicamentosusohumano- 3/la-aemps-autoriza-el-car-t-ari-0001-del-hospital-clinic-para-
pacientes- con-leucemia-linfoblastica-aguda/?lang=en. Accessed 13/03/2021.
E. McGrath and P. Machalik

193
Role ofAcademia
Academia remains very active in the early phases of clinical trials designed to evalu-
ate innovative GTMPs as potential complements, substitutes, or bridges to historical
forms of haematopoietic cell transplants. One recent study calculated that even now,
when industry interest in these therapies has increased signicantly in the last
5–6years, over 50% of CAR-T cell trials in the USA are still sponsored by aca-
demia (Kassir et al. 2020). Many public institutions have invested signicant
resources to upgrade their processing facilities to GMP-compliant levels, thus
allowing for small-scale manufacturing of experimental medicinal products to sup-
port phase I and possibly phase II studies. Furthermore, academia must become a
proactive stakeholder in the regulatory area by engaging with the authorities, shar-
ing their know-how and voicing their opinion. Through continental registries, such
as EBMT, academic institutions will continue to play a key role due to their data and
procedural knowledge, which will be very useful not only for researchers but also
for industry, health care regulators and payers (Hildebrandt 2020).
Health Technology Assessment
For a marketing authorization holder, approval by EMA is just one step. To gain
market access in the EU, the manufacturer must now approach national health care
reimbursement authorities, collectively known as health technology assessment
(HTA) bodies, who will assess the cost of the added value of novel therapies com-
pared to the current standard of care. Unlike the centralized authorization process,
HTA assessments are performed at the national level and are subject to great vari-
ability between member states. Over the past decade, the EU has pursued a more
harmonized HTA process across Europe, although there remains signicant resis-
tance among member states, and a legislative proposal adopted by the Commission
in early 2018 is only very slowly progressing through the parliamentary process.
Future Focus
Access to ATMPs, including cellular therapies, is likely to be a particular challenge
for patients, health care professionals, and national health systems due to their
expected high costs and complexity. Foreseeable changes to the regulatory frame-
work could see closer alignment with MA and HTA to make them more concurrent
and less sequential processes. The EMA’s strategy for big data places an emphasis
on using real-world data (RWD) to support regulatory decisions, and signicant
efforts are being made to prepare the structures to support this move. Accelerated
processes, such as PRIME, will continue to evolve as regulators gain more knowl-
edge and the science and medicine develop. The interplay between European
medicinal product regulations and genetically modied organism (GMO) frame-
works will likely continue to be the focus of efforts to harmonize interpretations
37 The Regulatory Framework for CAR-T Cells in Europe: Current Status…

194
across the EU.Regulators will see more automation of manufacturing processes,
which should help reduce risk and variability, while decentralized or ‘bedside’ man-
ufacturing could become more common but still need regulatory approval and over-
sight. Allogeneic CAR-T products will also require substantial evidence to reassure
regulators regarding safety concerns about graft versus host disease, cell rejection,
and the risks associated with gene editing. Data protection measures under the
General Data Protection Regulation (GDPR) for health-related personal data could
see adaptations to better facilitate secondary use of data collected to support inves-
tigational and regulatory needs.
Centre Qualification by Competent Authorities
andManufacturers
Shared Goals
A high degree of competencies is required from centres involved in CAR-T cell
therapies by both the competent authorities and the manufacturers. With regard to
centre qualication, authorities and manufacturers share at least some own goals,
which is minimizing CAR-T cell therapy-associated risks for patients to deliver safe
and efcient therapy. Authorities at the international, national, or regional levels
assess the quality of care, level of practice and health outcomes, and qualify centres
that successfully demonstrate high standards of health care and patient safety. For a
manufacturer, accreditation by the competent authority veries that the required
standards are followed and the necessary qualications, processes and resources are
present. From the centre’s perspective, receiving necessary accreditations and
approvals from both the competent authority and the respective manufacturer is a
prerequisite to support concrete CAR-T cell therapy.
Centre Assessment
The presence of accreditation by a competent authority is among the rst items
checked by a manufacturer during the so-called feasibility assessment. The other
reviewed items include the scope of authorized activities; the centre’s ability to
perform particular procedures and tests, incorporate the manufacturer’s require-
ments, and guarantee specic environmental conditions; and the presence of
requested equipment and qualied personnel. For a manufacturer, the assessment is
a great chance to obtain a better understanding of a centre’s setup and daily routine
and its procedural and capacity constraints. The assessment might also reveal gaps,
such as the inadequateness or complete absence of required processes. Generally,
the feasibility assessment is a unique opportunity to evaluate prospective candidates
for collaboration, and it precedes all other steps in a centre’s qualication by a
manufacturer, which may vary in scope and detail depending on the particular
therapy.
E. McGrath and P. Machalik

195
Centre Auditing
The centre qualication audit is usually performed by a manufacturer prior to com-
mencement of any collaboration. The aim is to evaluate compliance with applicable
regulatory requirements and the centre’s own procedures or policies. Manufacturers
obtain an appropriate understanding of the performed services and the robustness of
engaged systems, including quality management, personnel training, and the capac-
ity of available resources. They usually request some of the centre’s internal docu-
ments and process details to be shared prior to the audit to allow for a thorough
review. During the audit, auditors examine more of the centre’s documentation,
interview personnel, inspect the facility’s key locations, and evaluate processes in
targeted functional areas.
In the course of an established collaboration, other types of audits can be orga-
nized. The so-called surveillance audit is a periodic audit to ensure that a centre is
continuing to comply with the required standards. The emphasis is on reviewing
signicant changes that have occurred in the relevant procedures, facility and its
quality system since the qualication audit. A follow-up on any previous audit nd-
ings, including the implementation of corrective and preventive actions, is also a
common part of surveillance audits, which are usually performed every 2–3years.
A for-cause audit can be called in response to serious circumstances, including
deciency in meeting regulatory requirements, occurrence of a major deviation,
repeated deviations, or the risk or occurrence of patient safety issues. This audit
generally focuses on identied nonconformities and areas of manufacturer concern.
Any type of audit will result in an audit report, which lists audit observations or
ndings that might be evaluated for signicance as minor, major, or critical. In
response to a nding, a centre’s own internal procedure usually mandates insurance
of a corrective action. This frequently means strengthening the existing processes or
creation of brand-new processes. Acceptable responses to audit ndings are required
to close an audit.
Centre Training
Audited centres are further qualied by the manufacturers for support of concrete
CAR-T cell therapy. Manufacturers usually do not aim to boost personnel’s general
knowledge or the skills and attitudes required for daily routine practice. Their focus
is on explaining the specicities of clinical trials or authorized therapies, with few
differences between the requirements of the two categories. Generally, the critical
parameters of procedures and products are explained, as well as timelines, environ-
mental conditions, types of equipment and material, completion and usage of
involved documents, and principles of communication among stakeholders. Due to
the complexity of CAR-T cell therapies, centre personnel qualied by manufactur-
ers perform a wide range of functions. Manufacturers usually train those involved in
patient or donor care; starting material procurement, processing, intermediate stor-
age, packaging, release, and testing; completion of documents; and ATMP receipt,
37 The Regulatory Framework for CAR-T Cells in Europe: Current Status…

196
storage, thawing and administration. In practice, these functions involve physicians,
nurses, pharmacists, apheresis and laboratory technicians, and administrative
workers.
Required procedure parameters include duration limits, processed volume tar-
gets, type of anticoagulant, environmental conditions (such as temperature and
humidity), and methods of disconnecting and sealing collection bags. Product
parameters that manufacturers like to specify include targets for collected volume,
yield and purity and the required number of units and samples. With regard to time-
lines, the importance of procedure scheduling and harmonization with other proce-
dural steps or treatment sessions is emphasized. Manufacturers are usually very
clear about the type of equipment and material required for procurement, intermedi-
ate storage, indoor transport, the processing and packaging of starting material or
storage, and thawing and administration of ATMPs. The purpose and usage of the
involved documents are explained, and instructions for completing, archiving, or
sharing with other stakeholders are provided. Colour-coded sample documents, pre-
populated forms, and checklists are among the most frequent support materials.
Centre qualication is not bound to its on-site execution. It can also be per-
formed remotely when travel or visitor restrictions or social distancing guidelines
make any externally driven on-site activities impossible. Internet-based applica-
tions, teleconferencing tools, and purposely developed virtual procedures have
recently been successfully used by manufacturers to perform feasibility assess-
ments, audits, and trainings.
Key Points
• CAR-T cell therapy falls under the ATMP framework, presenting chal-
lenges to all stakeholders, including health care providers and patients.
• Regulatory issues concern not only marketing authorization but also mech-
anisms for cost–benet assessment and, less directly, GMOs and data
protection.
• Academia will continue to play a signicant role in the development and
delivery of these new therapies and should expect to engage with other
stakeholders.
• The regulatory framework is not static and evolves with experience and
knowledge.
• Competent authorities and manufacturers have a common goal, which is
minimizing CAR-T cell therapy-associated risks for patients.
• Accreditation of a centre by a competent authority is understood as veri-
cation that the required standards are followed and the necessary qualica-
tions, processes and resources are present.
• Manufacturers qualify centres for support of a concrete CAR-T cell ther-
apy and focus on the specicities of a particular project.
• Centre qualication is not bound to on-site execution and can be performed
remotely using internet-based applications, teleconferencing tools, and
purposely developed virtual procedures.
E. McGrath and P. Machalik

197
Sources of Information
• World Health Organization—Patient Safety [Internet]. 13 September 2019.
Available from: https://www.who.int/news- room/fact- sheets/detail/patient-
safety (Accessed February 2021)
• Yakoub-Agha I, Chabannon C, Bader P, Basak GW, Bonig H, Ciceri F,
Corbacioglu S, Duarte RF, Einsele H, Hudecek M, Kernsten MJ, Köhl U, Kuball
J, Mielke S, Mohty M, Murray J, Nagler A, Robinson S, Saccardi R, Sanchez-
Guijo F, Snowden JA, Srour M, Styczynski J, Urbano-Ispizua A, Hayden PJ,
Kröger N. Management of adults and children undergoing chimeric antigen
receptor T-cell therapy: best practice recommendations of the European Society
for Blood and Marrow Transplantation (EBMT) and the Joint Accreditation
Committee of ISCT and EBMT (JACIE) [Internet]. February 2020. Available
from: https://haematologica.org/article/view/9515 (Accessed February 2021)
• Taylor L, Rodriguez ES, Reese A, Anderson K.Building a Program—Implications
for infrastructure, nursing education, and training for CAR-T cell therapy. Clin J
Oncol Nurs. 2019 Apr 1;23(2):20–26. https://doi.org/10.1188/19.CJON.
S1.20- 26. [Internet]. Available from: https://pubmed.ncbi.nlm.nih.gov/30880820/
(Accessed February 2021)
• Black A, Gabriel S, Cauleld D.Implementing chimeric antigen receptor T-cell
therapy in practice. The Pharmaceutical Journal [Internet]. 22 May 2020.
Available from: https://pharmaceutical- journal.com/article/ld/implementing-
chimeric- antigen- receptor- t- cell- therapy- in- practice (Accessed February 2021)
• Quality System Audit Program—Activate apheresis and cell collection sites
faster by licensing GTP audit results. Be The Match BioTherapies. [Internet].
Available from: https://bethematchbiotherapies.com/solutions/quality- -
system- audit- program/ (Accessed February 2021)
• Vincinis R. Major vs. Minor Audit Findings. The Auditor. [Internet].
Available from: https://www.theauditoronline.com/major- vs- minor- audit-
ndings/ (Accessed February 2021)
References
Detela G, Lodge A.EU regulatory pathways for ATMPs: standard, accelerated and adaptive path-
ways to marketing authorisation. Mol Therap Method Clin Develop. 2019;13:205–32. https://
doi.org/10.1016/j.omtm.2019.01.010.
Hildebrandt M. Horses for courses: an approach to the qualication of clinical trial sites and
investigators in ATMPs. Drug Discov Today. 2020;25(2):265–8. https://doi.org/10.1016/j.
drudis.2019.10.003.
Kassir Z, etal. Sponsorship and funding for gene therapy trials in the United States. JAMA Am
Med Assoc. 2020;323(9):890. https://doi.org/10.1001/jama.2019.22214.
McGrath E, Chabannon C.Regulatory aspects of ATMP versus minimally manipulated immune
cells. In: The EBMT handbook: hematopoietic stem cell transplantation and cellular therapies.
Springer; 2018. p.461–4. https://doi.org/10.1007/978- 3- 030- 02278- 5_62.
37 The Regulatory Framework for CAR-T Cells in Europe: Current Status…

198
Open Access
This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
E. McGrath and P. Machalik

199
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_38
R. Saccardi
Cell Therapy and Transfusion Medicine Unit, Careggi University Hospital, Florence, Italy
e-mail: [email protected].it
F. Sanchez-Guijo (
*)
Cell Therapy Area and Hematology Department, University Hospital of Salamanca,
Salamanca, Spain
Department of Medicine, University of Salamanca, Salamanca, Spain
e-mail: [email protected]
38
How Can Accreditation Bodies, Such
asJACIE or FACT, Support Centres
inGetting Qualified?
RiccardoSaccardi andFerminSanchez-Guijo
The FACT-JACIE accreditation system is based on a standard-driven process cover-
ing all the steps of HSC transplant activity, from donor selection to clinical care.
Since the rst approval of the First Edition of the Standards in 1998, over 360 HSCT
programmes or facilities have been accredited at least once, most of them achieving
subsequent re-accreditations (Snowden et al. 2017). The positive impact of the
accreditation process in the EBMT Registry has been well established (Gratwohl
etal. 2014). Starting with version 6.1, the standards include new items specically
developed for other cellular therapy products, with special reference to immune
effector cells (IECs). This reects the rapid evolution of the eld of cellular therapy,
primarily (but not exclusively) through the use of genetically modied cells, such as
CAR-T cells. FACT-JACIE standards cover a wide range of important aspects that
can be of use for centres that aim to be accredited in their countries to provide IEC
therapy. Notably, FACT-JACIE accreditation itself is a key (or even a prerequisite)
condition in some countries for approval by health authorities to provide commer-
cial CAR-T cell therapy and is also valued by pharmaceutical companies (both
those developing clinical trials and those manufacturing commercial products),
which also inspect the cell therapy programmes and facilities established at each
centre (Hayden etal. 2021). Interest in applying for FACT-JACIE accreditation that
includes IEC therapeutic programmes is clearly increasing, from four applications
in 2017 to 36 applications approved in 2019. The standards do not cover the manu-
facturing of such cells but include the chain of responsibilities when the product is

200
provided by a third party (Maus and Nikiforow 2017). In any case, all the steps in
the process in which the centre is involved (e.g., patient or donor evaluations, cell
collection, cell reception, and storage) are covered by the standards, including the
appropriate agreements with the internal partners, including the pharmacy depart-
ment. In addition, from a clinical perspective, IECs may require special safety mon-
itoring systems due to the high frequency of acute adverse events related to the
massive immunological reaction against the tumour. Although examples and expla-
nations are found in the standard manual, here, the special importance of identifying
and managing cytokine release syndrome (CRS) should be emphasized, and the
standards focus not on specic therapeutic algorithms but on ensuring that medical
and nursing teams are sufciently trained in the early detection of this and other
potential complications (e.g., neurological complications). They also pay attention
to the full-time availability within the institution and its pharmacy of the necessary
medication to address complications and the capacitation and involvement of
Intensive Care and Neurology Department professionals to provide urgent care if
needed. Forthcoming cellular therapy products, currently under investigation, will
show a wider range of risk proles, therefore requiring product-specic risk assess-
ment and consequent adaptation of the clinical procedures for different classes of
products. The FACT-JACIE standards will continue to adapt to these future needs to
assist centres in their achievement of optimal clinical outcomes.
References
Gratwohl A, Brand R, McGrath E, etal. Use of the quality management system “JACIE” and out-
come after hematopoietic stem cell transplantation. Haematologica. 2014;99:908–15.
Hayden PJ, Roddie C, Bader P, Basak GW, Bonig H, Bonini C, et al. Management of adults and
children receiving CAR T-cell therapy: 2021 best practice recommendations of the European
Society for Blood and Marrow Transplantation (EBMT) and the Joint Accreditation Committee
of ISCT and EBMT (JACIE) and the European Haematology Association (EHA). Ann Oncol.
2021;S0923–7534(21):04876–6. https://doi.org/10.1016/j.annonc.2021.12.003. Online ahead
of print.
Maus MV, Nikiforow S.The why, what, and how of the new FACT standards for immune effector
cells. J Immunother Cancer. 2017;5:36.
Snowden JA, McGrath E, Duarte RF, etal. JACIE accreditation for blood and marrow transplan-
tation: past, present and future directions of an international model for healthcare quality
improvement. Bone Marrow Transplant. 2017;52:1367–71.
Key Points
• FACT-JACIE standards have helped accredited centres improve their
HSCT clinical outcomes for more than 20years.
• Standards have been adapted to cover immune effector cell (IEC) therapy
and are a key element in demonstrating optimal quality and performance
for seeking accreditation by both National Health Authorities and the phar-
maceutical companies involved.
• The IEC product chain of responsibilities, agreements with all involved
partners, and full coverage of related adverse events are among the key
elements of IEC- related standards.
R. Saccardi and F. Sanchez-Guijo

201
Open Access
This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
38 How Can Accreditation Bodies, Such as JACIE or FACT, Support Centres…

203
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_39
N. Kröger (*)
Department of Stem Cell Transplantation, University Medical Center Hamburg-Eppendorf,
Hamburg, Germany
e-mail: [email protected]
J. Gribben
Bart’s Cancer Institute, Queen Mary University of London, London, UK
e-mail: [email protected]
I. Sánchez-Ortega
EBMT, Executive Ofce, Barcelona, Spain
e-mail: [email protected]g
39
Educational Needs forPhysicians
NicolausKröger
, JohnGribben
, andIsabelSánchez-Ortega
CAR-T cells are novel therapies associated with promising and potentially curative
outcomes in patients with high-risk relapsed disease. In Europe, there are currently
three approved products (tisagenlecleucel, axicabtagene ciloleucel, and brexucabta-
gene autoleucel) for patients with acute lymphoblastic leukaemia, aggressive B-cell
lymphoma, and mantle cell lymphoma, although expanded haematologic and non-
haematologic indications are expected soon.
Cellular therapy, including CAR-T cells, is a rapidly evolving eld in haematol-
ogy, and treatment is becoming personalized and specic. To ensure optimal
decision- making by physicians, adequate education programmes must be available
and must be regularly updated. There is a need to identify knowledge gaps and bar-
riers to address these issues with continuous medical education. Adequate education
increases the competence and performance of physicians and improves the quality
of decision-making, ultimately resulting in the optimization of patient management.
The importance of education is also reected in the JACIE accreditation scheme, the
major objective of which is to promote quality medical and laboratory practice in
cellular therapy by offering accreditation based on internationally recognized stan-
dards. The relevant standards in this scheme require that clinical, collection, and
processing facility staff participate in continuous education activities (JACIE 2021).
However, there is also a need to educate the wider community (people who do not

204
work at JACIE accredited sites) to ensure sufcient knowledge to recognize the role
of CAR-T therapy, identify suitable patients and understand the process for timely
referral to treatment centres. There is an inevitable delay between referral, cell col-
lection, and delivery of the CAR-T products, and physicians must be aware of this
process and take steps to manage their patients, who are at high risk of rapid disease
progression and may require bridging therapy, ideally in close collaboration with
the CAR-T treatment centre. Therefore, referring physicians must be educated to
understand the patient selection process, T cell collection process, and the process-
ing and conditioning therapy to fully understand the path that their patients will
travel and the time frames involved in delivering these complex treatments.
CAR-T cell therapies are associated with remarkable therapeutic response rates
but also with unique and potentially lethal complications that require specic edu-
cational updates. Cytokine-release syndrome (CRS) and neurotoxicity are the most
frequent complications after CAR-T cell therapies. These complications can occur
concomitantly and may have a very rapid onset, with a spectrum of symptoms that
range from mild to life threatening. In addition, CRS onset is often indistinguish-
able from infection, which, in the setting of neutropenia, makes the management of
these complex patients even more challenging (Hayden etal. 2021). Haematologic
toxicity, most often seen as a complication of lymphodepleting induction therapy, is
frequent after CAR-T cell infusion, but the pattern, duration, and outcome are not
well described. Learning to monitor and adequately treat persistent cytopenias is
necessary for adequate management of these patients. Learning to dene the opti-
mal timing for ICU referral is also critical because any delay in ICU admission can
compromise patient outcomes. In addition, the unique toxicity prole of CAR-T
cell therapies makes incorporation of real-life data, including that from the patients’
perspective, essential, and initial data suggest that patient-reported toxicities and
mental health concerns are common throughout all stages of survivorship (Barata
etal. 2021; Hoogland etal. 2021). From the moment that a patient is identied as a
CAR-T candidate, education and supportive care of patients undergoing CAR-T
therapy is crucial to improve the knowledge and experience of the patients and their
families. To address these issues, a trained multidisciplinary team, including haema-
tologists, oncologists, intensivists, neurologists, pharmacists, psychologists, and
nurses, must work together from the time of potential patient identication to the
time of treatment and discharge, and their roles are crucial at different stages in the
CAR-T cell process.
Large registry studies with high-quality data may provide the basis of knowledge
for CAR-T cell therapies and open the door to the necessary specic subpopulation
investigations. To ensure continuous evaluation of the efcacy and safety of com-
mercially available CAR-T cells, the EMA endorsed the use of the EBMT registry
for collection of 15-year follow-up data of treated patients (EMA 2019). Likewise,
follow-up data of patients receiving academic and other pharmaceutical-sponsored
CAR-T cell therapies are also expected to be reported to the EBMT registry.
Therefore, the real-world data contained in the EBMT registry will likely be a major
source of knowledge to improve the use of CAR-T cell therapies and to understand
the short-term and long-term patient toxicities and outcomes. This will also allow us
N. Kröger et al.

205
to gain insights into potential biomarkers and the patient and disease characteristics
that might impact the efcacy of CAR-T-treatment, opening the path to more effec-
tive selection and stratication of patients.
Ongoing investigations of CAR-T cell therapies are seeking to elucidate the
mechanisms of resistance, immune escape, and relapse so that the current barriers
can be overcome and treatment efcacy can be improved. Research is also focused
on access to “off-the-shelf” allogeneic CAR-T products, simplifying the manufac-
turing process and mitigating side effects, among other aims. Thus, the complexity
and rapid changes in the eld of cellular therapies demands wide collaboration to
maintain up-to-date education on the entire pathway from collection to the manu-
facturer and back to the clinical unit. GoCART, a multistakeholder coalition
launched by EBMT and EHA, offers a platform to provide the required diversied
and topic-specic education on CAR-T cell therapies. Likewise, the annual EBMT/
EHA European CAR-T cell meeting provides specic continuous medical educa-
tion in this complex eld. In addition, educational online updates are provided on
the EBMT and EHA e-learning platforms (https://www.ebmt.org/education/e- -
learning, https://ehacampus.ehaweb.org) with specic webinars and e-learning
courses focused not only on CAR-T cells but also on other evolving immunotherapy
treatments that may impact the pathway towards CAR-T cell treatment. There is
still much to learn, and this rapidly evolving eld requires rapid and constant edu-
cational updates.
References
Barata A, Hoogland A, Hyland K, et al. Patient-reported toxicities in axicabtagene ciloleucel
recipients: 1-year follow-up. Transpl Cell Therap. 2021;27(3):S375. https://doi.org/10.1016/
S2666- 6367(21)00485- 1.
EMA qualication opinion on cellular therapy module of the EBMT Registry. 2019. Available
at: https://www.ema.europa.eu/en/cellular- therapy- module- european- society- blood- marrow-
transplantation- ebmt- registry.
Key Points
• Continuous medical education should ll unavoidable knowledge gaps in
a rapidly evolving eld.
• Big data registry studies, multistakeholder coalitions, and multidisci-
plinary educational meetings provide regular updates on the entire CAR-T
cell therapy process.
• Updates on specic topics and the latest scientic developments are also
required to provide individualized high-quality patient management.
• e-learning platforms and CAR-T cell meetings provide adequate and spe-
cic updates in this complex eld, but there is also a need to educate the
wider medical community, who refer patients to treatment centres.
• Continuous medical education is necessary, especially because this eld is
rapidly evolving.
39 Educational Needs forPhysicians

206
Hayden PJ, Roddie C, Bader P, Basak GW, Bonig H, Bonini C, etal. Management of adults and
children receiving CAR T-cell therapy: 2021 best practice recommendations of the European
Society for Blood and Marrow Transplantation (EBMT) and the Joint Accreditation Committee
of ISCT and EBMT (JACIE) and the European Haematology Association (EHA). Ann Oncol.
2021;S0923–7534(21):04876–6. https://doi.org/10.1016/j.annonc.2021.12.003. Online ahead
of print.
Hoogland A, Jayani R, Collier A, etal. Acute patient-reported outcomes in B-cell malignancies
treated with axicabtagene ciloleucel. Cancer Med. 2021;10(6):1936–43.
JACIE. 2021. Available at: https://www.ebmt.org/accreditation/jacie- standards.
Open Access
This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
N. Kröger et al.

207
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_40
M. Kenyon (*)
Department of Haematological Medicine, King’s College Hospital, NHS Foundation Trust,
London, UK
e-mail: [email protected]
J. Murray
Haematology and Transplant Unit, The Christie NHS Foundation Trust, Manchester, UK
e-mail: [email protected]
R. Ellard
The Royal Marsden NHS Foundation Trust, London, UK
e-mail: [email protected]
D. Hutt
Department of Pediatric Hematology-Oncology and BMT, Edmond and Lily Safra Children’s
Hospital, Sheba Medical Center, Ramat Gan, Israel
e-mail: [email protected].il
40
Education Needs forNurses inAdult
andPaediatric Units
MichelleKenyon, JohnMurray, RoseEllard, andDaphnaHutt
Complex nursing care for patients on the CAR-T cell therapy pathway involves
many different nursing roles that have important functions at different stages in the
pathway. Within the multiprofessional team, nurse education is critical to safe and
competent care and to the patient’s treatment experience. As we consider the educa-
tion needs of the nursing workforce throughout the entire patient pathway, including
the supply chain, chain of custody, and clinical care delivery, we recognize the
important roles of expert nurses, practice educators, and the wider multiprofessional
team in sharing their knowledge and experience. Nurse education strategies should
include referring nursing teams to facilitate seamless patient care throughout refer-
ral, treatment, and follow-up to optimize communication and appropriately meet
patient and caregiver information needs.
Treatment plans can change rapidly; patients do not reach the point of treatment
or relapse during admission. The involvement of disciplines such as palliative care
and psychological therapy in the programme is key. The relationship between the

208
referring and treating centre is critical, and an active dialogue between teams from
the time of referral is imperative to optimize patient care.
Apheresis andCell Collection
Nurses with the knowledge, skills, and expertise to perform CAR-T-specic apher-
esis procedures do so in many JACIE accredited centres following training and
competency achievement. Apheresis booking is synchronized with the availability
of manufacturing space in the pharmaceutical company, but timing is critical to
maximize collection quality and minimize the risk of unsalvageable disease pro-
gression. Preprocedure work-up can include disease- and product-specic tests and
screening. Technically, apheresis is similar to donor lymphocyte or mononuclear
cell procedures but may be more challenging due to low lymphocyte counts follow-
ing earlier treatments. The patients may be symptomatic due to the disease burden
and previous therapies and can become unwell during harvest.
Cell Infusion
Thawing and cell infusion are performed in most centres by appropriately trained
nurses. Frozen cells are shipped from the manufacturer, and thawing occurs at the
bedside via a water bath or automated device. Specic training on defrosting and
infusing the product is mandatory. Soft waste will be disposed of into a double
clinical waste bag, tagged, numbered, and placed in a dedicated biohazard waste
container. Sharp waste, e.g., syringes and vials, must be placed in sealed and lidded
sharps container, which is tagged and labelled as biohazard waste. A disposal
record should be maintained. PPE should be worn at all times of disposal. If cloth-
ing becomes contaminated, it should be changed immediately and disposed of as
soft waste. If spillage occurs, the spill should be cleaned while wearing PPE and
using Clinell red wipes or other virucidal products. Routine checks are performed
at the bedside (patient ID, consent, prescription, vital signs, IV access).
Premedication is administered, ensuring that no steroids are given. The cells are
infused as per local policy and the product specication. The patient’s vital signs
are recorded during and following the infusion. All necessary documentation is
completed. The infusion is usually uneventful, but intensive care and neurology
services should be notied of the CAR-T infusion should their support be needed
during the postinfusion period.
Patient Monitoring
The two most common toxicities following CAR-T infusion are cytokine release
syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome
(ICANS).
M. Kenyon et al.

209
CRS is the most common acute toxicity. Frequently reported symptoms are
fever, hypoxia, and hypotension, which can mimic neutropenic sepsis. Thus, the
patient must be treated for suspected infection with intravenous antibiotics and a
full septic screen must be performed.
ICANS symptoms can be progressive and may include aphasia, an altered level
of consciousness, impairment of cognitive skills, motor weakness, seizures, and
cerebral oedema (Lee etal., 2019). Nurses should be aware of these symptoms, and
familiarity with the patient’s baseline condition aids in monitoring for subtle
changes.
Vital signs of inpatients should be recorded at least once every 4 h to monitor
for signs/symptoms of CRS.Patients may deteriorate quickly, and nurses should
promptly report concerns to the medical team to ensure early recognition and
treatment. The recommended monitoring and assessment tools for CRS and
ICANS are the American Society of Transplant and Cellular Therapy CRS con-
sensus grading (Table1in Chap. 26), immune effector cell-associated encepha-
lopathy (ICE) tool (Table 1 in Chap. 27), and the ASTCT ICANS consensus
grading (Table2in Chap. 27) (Lee etal., 2019). The CRS grade should be calcu-
lated if there is a deterioration in the patient’s vital signs and reported to the medi-
cal team. The ICE score should be calculated at least twice daily. This tool is of
particular benet, as subtle handwriting changes can be an early sign of ICANS.If
the ICE score is less than 10, the ASTCT ICANS grade (Table3 in Chap. 27)
should be calculated and the medical team notied of the change in the patient’s
condition. Patients require daily blood tests, including full blood count, biochem-
istry, CRP, and ferritin; some centres may have additional routine tests.
Toxicity Management
Treatment of symptoms is a key nursing role in the management of CAR-T toxici-
ties. Patients with suspected CRS may require supportive measures, such as
paracetamol, IV uids, or supplemental oxygen. The rst-line medicinal treatment
for CRS is tocilizumab, an anti-IL6 monoclonal antibody given intravenously. Up to
four doses can be given, at least 8 h apart. Second-line treatment for CRS is usually
corticosteroids, although these are always used with caution due to the potential
deleterious effect on CAR-T cell efcacy. However, ICANS is typically treated with
corticosteroids as a rst line because tocilizumab is a large molecule and does not
cross the blood–brain barrier.
Discharge
Upon discharge, patients and their caregivers should have written information about
potential side effects and who and how to contact an appropriate CAR-T member if
they develop problems or concerns. Patients must be aware of the symptoms of CRS
and serious neurological reactions and the need to report all symptoms to the CAR-T
40 Education Needs forNurses inAdult andPaediatric Units

210
team immediately. If discharged prior to day 28, patients are required to remain
within close proximity of the treatment centre until day 28. They are also advised
not to drive for 8 weeks post-infusion or resolution of neurologic symptoms due to
the risk of delayed neurotoxicity. Ideally, they should have a responsible adult as a
caregiver for the rst 3 months at home.
Long-Term Follow Up
In the CAR-T setting, the recommended minimum duration of follow-up is 15
years, with annual assessment, which fulls the regulatory requirements and allows
submission of longitudinal outcome data that can contribute to the growing evi-
dence base. The range of assessments and late effects screening can vary between
products and disease indications. Nurse awareness is necessary to support the
patient with appointments, coordination of tests, communication of results, and
escalation of patient concerns when raised. Early quality of life data show promis-
ing improvements (Tam et al. 2019) for some patients who achieve PR and
CR.Survivorship care, supporting the patient and caregiver through the transition
from treatment through recovery and beyond, is a key area for nurse development.
Paediatric Considerations
Currently, tisagenlecleucel (Kymriah™) is the only approved treatment for refrac-
tory/relapsed ALL in children and young adults up to 25 years of age. Apheresis in
small children is considered safe but challenging because it has potentially more
side effects than in adults due to the small body mass and unique physiology of
children. Venous access in small children can be difcult and limits inlet rates and
in some cases requires insertion of a leukapheresis catheter (Mahadeo etal. 2019).
Children weighing 20–25kg may require priming of the machine with packed red
cells prior to the apheresis procedure. Metabolic complications due to citrate toxic-
ity may present differently in children (Del Fante etal. 2018). Obtaining a sufcient
number of harvested cells could be a limiting factor in infants and small children
(Hayden etal. 2021). In the pre-apheresis consultation, the nurse should consider all
of the above issues and provide age-appropriate preparation for the procedure,
including descriptions of the sequence of events that will occur and accurate infor-
mation on what pain and sensations to expect.
Hypotension and hypoxia are the principal determinants of the consensus grad-
ing scale, and hypotension assessment should account for age and the patient’s indi-
vidual baseline. Although the 10-point ICE assessment is useful for screening adults
for encephalopathy, its use in children may be limited to those aged ≥12 years with
sufcient cognitive ability to perform it. In children aged <12 years, the Cornell
Assessment of Pediatric Delirium (CAPD) is recommended to aid in the overall
grading of ICANS (Lee etal. 2019) (Table40.1).
M. Kenyon et al.

211
After treatment, children with B-cell aplasia should receive immunoglobulin
replacement to maintain IgG levels according to institutional guidelines for IgG
substitution (i.e., ≥500 mg/dL) (Hayden etal. 2021).
For patients aged 1–2 years, the following serve as guidelines for the correspond-
ing questions:
1. Holds gaze, prefers primary parent, looks at speaker.
2. Reaches and manipulates objects, tries to change position, if mobile may try
to get up.
3. Prefers primary parent, upset when separated from preferred caregivers.
Comforted by familiar objects (i.e., blanket or stuffed animal).
4. Uses single words or signs.
5. No sustained calm state.
6. Not soothed by usual comforting actions, e.g., singing, holding, talking, and
reading.
7. Little if any play, efforts to sit up, pull up, and if mobile crawl or walk around.
8. Not following simple directions. If verbal, not engaging in simple dialogue with
words or jargon.
Table 40.1 Encephalopathy assessment for children age <12 years using the CAPD
Answer the following based on interactions with the child over the course of the shift
Never, 4 Rarely, 3 Sometimes, 2 Often, 1 Always, 0
1. Does the child make eye
contact with the caregiver?
2. Are the child’s actions
purposeful?
3. Is the child aware of his or
her surroundings?
4. Does the child communicate
needs and wants?
Never, 0 Rarely, 1 Sometimes, 2 Often, 3 Always, 4
5. Is the child restless?
6. Is the child inconsolable?
7. Is the child underactive; very
little movement while awake?
8. Does it take the child a long
time to respond to
interactions?
Adapted from Traube etal. 2021; reproduced with permission
40 Education Needs forNurses inAdult andPaediatric Units

212
References
Del Fante C, Seghatchian J, Perotti C.Reections on methodical approaches to hematopoietic stem
cell collection in children. Transfus Apher Sci. 2018;57(3):425–7. https://doi.org/10.1016/j.
transci.2018.05.005.
Hayden PJ, Roddie C, Bader P, Basak GW, Bonig H, Bonini C, etal. Management of adults and
children receiving CAR T-cell therapy: 2021 best practice recommendations of the European
Society for Blood and Marrow Transplantation (EBMT) and the Joint Accreditation Committee
of ISCT and EBMT (JACIE) and the European Haematology Association (EHA). Ann Oncol.
2021;S0923–7534(21):04876–6. https://doi.org/10.1016/j.annonc.2021.12.003. Online ahead
of print.
Lee DW, Santomasso BD, Locke FL, Ghobadi A, Turtle CJ, Brudno JN, etal. ASTCT consen-
sus grading for cytokine release syndrome and neurologic toxicity associated with immune
effector cells. Biol Blood Marrow Transplant. 2019;25(4):625–38. https://doi.org/10.1016/j.
bbmt.2018.12.758.
Mahadeo KM, Khazal SJ, Abdel-Azim H, Fitzgerald JC, Taraseviciute A, Bollard CM, et al.
Management guidelines for paediatric patients receiving chimeric antigen receptor T cell ther-
apy. Nat Rev Clin Oncol. 2019;16(1):45–63.
Schmidts A, Wehrli M, Maus MV.Toward better understanding and management of CAR-T cell-
associated toxicity. Annu Rev Med. 2021;72:365–82.
Tam C, Waller E, Jaeger U, Pacaud L, Ma Q, Maziarz R. Prolonged improvement in patient
reported quality of life (QoL) following tisagenlecleucel infusion in adult patients (pts) with
relapsed/refractory (r/r) diffuse large B-cell lymphoma (DLBCL): 19-month follow-up (FU) of
the Juliet study. BBMT. 2019;25(3):181–2.
Traube C, Gerber LM, Mauer EA, Small K, Broglie L, Chopra YR, et al. Delirium in children
undergoing hematopoietic cell transplantation: a multi-institutional point prevalence study.
Front Oncol. 2021;11:627726.
Key Points
• Nurse education strategies should recognize the importance of the range of
nursing roles at various stages in the CAR-T patient pathway and their dif-
fering education and training needs.
• Treatment plans may not always proceed as expected, and patients can
experience sudden and signicant changes.
• Apheresis is technically similar to donor lymphocyte or mononuclear cell
procedures but may be more challenging due to low lymphocyte counts,
poor physical condition or high symptom burden.
• Specic training on defrosting and infusing the product is mandatory.
• The two most common toxicities following CAR-T infusion are cytokine
release syndrome (CRS) and immune effector cell-associated neurotoxic-
ity syndrome (ICANS), for which patients are very closely monitored.
• Nurses must be trained in the use of the CRS and ICANS assessment tools,
local escalation protocols, and treatment strategies.
• Specic considerations exist for paediatric patients, and these nurses must
be trained accordingly.
M. Kenyon et al.

213
Open Access
This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
40 Education Needs forNurses inAdult andPaediatric Units

215
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_41
M. Galassi
Centrale Produzione Farmaci, Hospital Pharmacy, National Cancer Institute of Milan,
Milan, Italy
e-mail: [email protected]
M. E. Moreno-Martínez (
*)
Department of Pharmacy, Hospital de la Santa Creu i Sant Pau, Barcelona, Spain
e-mail: [email protected]
41
Role ofPharmacists
MargheritaGalassi andMariaEstelaMoreno-Martínez
The pharmacist has a key role in the management of CAR-T therapies. Selection,
ordering, reception, storage, preparation of the product for infusion, and dispensing
of CAR-T therapies are some of the pharmacy service responsibilities (Black 2018;
Moreno-Martínez etal. 2020; Booth etal. 2020). The pharmacist requires specic
training, ensuring coordination with all the professionals in the multidisciplinary
team who are involved in the management of these therapies, as summarized in
Table41.1.
The pharmacist must also know which types of CARs are available and can
arrive in the future. CAR-T cells are currently indicated for the treatment of B-cell
acute lymphoblastic leukaemia and diffuse large B-cell lymphoma, two haemato-
logical diseases that share expression of the CD-19 antigen, but the target antigens
are potentially many; therefore, the pharmacist must receive training that takes into
account new future possibilities. An example of other options is the advanced phase
experimentation of CAR-T cells and anti-B-cell maturation antigen (BCMA) for
multiple myeloma pathologies.
CAR-T cells are just the beginning, and CAR-Technology is being applied to
other immune cells:
• CAR natural killer (NK) cells: CAR-NK.
• CAR macrophages (M): CAR–M.

216
Finally, CAR-T cells may be effective against solid tumours, and the main prob-
lem related to the accessibility of the antigen can be solved in patients suffering
from glioblastoma and neuroblastoma with the injection of the cells on site.
The complexity of these therapies requires the intervention of the pharmacist
whose training must include implementation and management of advanced biotech-
nological procedures; therefore, specic skills not only in the preparation of classic
chemotherapies and monoclonal antibodies but also in how to handle, store, and
manage novel therapies and the specic medical devices that could be required
are needed.
One of the most important pharmacist interventions is patient follow-up, intended
to monitor toxicities, adverse events, and concomitant and contraindicated drugs.
Cytokine release syndrome is an extremely serious event that must be monitored by
a multidisciplinary team in which the pharmacist is the reference gure for the man-
agement of rescue drugs and pharmacovigilance studies.
Table 41.1 Pharmacist’s responsibilities
Pharmacist-specic training
Selection and indication for CAR-T cell therapy
• Review and approval for formulary addition
• Patient eligibility criteria
Ordering: know each procedure to order the drug
Reception
• Check integrity of the product, labelling, and temperature compliance
• Check certicate of analysis
Storage and handling
• Manage products stored at ultra-cold temperatures
• Action plan if temperature deviation
Dispensing
• Validate lymphodepleting chemotherapy and coordinate date of dispensing and time planned
for infusion
• Ensure chain of identity of cell product
• Check defrosting procedure. Record the date and time of all the procedures
Administration: conrm procedure and doses of tocilizumab stock ready to use
Follow-up
• Drugs permitted and contraindicated
• Monitoring and management of toxicities
• Ensure appropriate treatment is available
Patient and staff education
Key Points
• The pharmacist requires specic training in the management of CAR-T
therapies, ensuring coordination with all the professionals in the multidis-
ciplinary team.
• The pharmacist must know the types of CARs available and what will
arrive quite soon.
• New skills are needed to handle and store CARs and to follow-up with
patients.
M. Galassi and M. E. Moreno-Martínez

217
References
Black A.Pharmacy institutional readiness for marketed CAR-T therapy: checklists for pharmacy
services. Version 3.0. Specialist Pharmacy Service; 2018. Available at: https://www.sps.nhs.uk/
wp- content/uploads/2018/10/FINAL- Pharmacy- Institutional- Readiness- for- Marketed- CAR-
TDec- 2018.pdf
Booth JP, Kusoski CL, Kennerly-Shah JM.The pharmacist’s role in chimeric antigen receptor T
cell therapy. J Oncol Pharm Pract. 2020;26(7):1725–31.
Moreno-Martínez ME, Vinent-Genestar J, Muñoz-Sánchez C, Carreras-Soler MJ.Hospital phar-
macist’s roles and responsibilities with CAR-T medicines. Farm Hosp. 2020;44(1):26–31.
Open Access
This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
41 Role ofPharmacists

219
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_42
B. Calmels (*)
Cell Therapy Facility, Institut Paoli-Calmettes, Marseilles, France
e-mail: [email protected].fr
42
Educational Needs forCell Processing
Facility Personnel
BorisCalmels
Training on ATMP processes and procedures is systematically provided by the
sponsor after site certication/accreditation.
Chain of Identity (COI) and Chain of Custody (COC) are the most crucial items
to understand to ensure traceability and identication of a cellular product; for com-
mercial ATMPs, the COI and COC are usually managed through a dedicated secure
web-based platform.
In the autologous setting, cell processing staff is involved in most of the on-site
ATMP stages, from collection to administration: the information ow between all
protagonists must be well established to allow for timely delivery of information.
Pivotal training steps for cell processing staff:
– before apheresis: receipt of empty shipper (if apheresis is shipped after cryo-
preservation) and materials.
– after apheresis completion: control of product label, sampling, cryopreservation,
and/or packaging for transportation of fresh or frozen apheresis to the manufac-
turing site.
The responsibility for subsequent steps might be shared with or fullled by the
hospital pharmacy, depending on local regulations.
– after manufacturing completion and prior to initiating lymphodepletion: receipt
of shipper, conformity of transport temperature, frozen bag integrity, and transfer
of frozen product to on-site storage.
– the day of infusion: transport of bag(s) to the thawing site, clinical ward (if bed-
side), or cell therapy facility (recommended) if localized near the infusion site
(thawed ATMPs need to be infused asap).

220
Frozen bag handling and thawing require expertise and must be performed by
experienced, i.e., cell processing staff, whenever possible: this will also relieve
training of unexperienced staff, especially regarding the risks associated with liquid
nitrogen exposure, and anoxia or handling of accidental bag failures.
One of the many challenges of training is to become used to the quantity and
variety of forms associated with each step of the ATMP circuit; consequently, mock
(training) runs organized by sponsors are pivotal for staff training.
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
Key Points
• Chain of Custody (COC) is crucial to ensure traceability through the mul-
tiple steps and stakeholders in the supply chain.
• Information that ows between all protagonists must be well dened.
• Frozen bag handling and thawing should be performed by skilled cell pro-
cessing staff.
B. Calmels

221
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_43
S. R. Terwel (*)
EBMT GoCART, Leiden, The Netherlands
e-mail: GoCAR[email protected]
J. Kuball
Department of Hematology, and Center for Translational Immunology, University Medical
Center Utrecht, Utrecht, The Netherlands
Legal Regulatory Affairs Committee (LRAC) of EBMT, Barcelona, Spain
e-mail: [email protected]
M. Dreyling
Department of Medicine III, Ludwig Maximilians University Munich, Munich, Germany
e-mail: [email protected]
F. Cerisoli
EHA, The Hague, The Netherlands
e-mail: f.cerisoli@ehaweb.org
43
GoC ART
SofieR.Terwel, JürgenKuball, MartinDreyling,
andFrancescoCerisoli
Cellular therapies manufactured from cells of haematopoietic origin, such as
CAR-T–cell therapies, provide a revolutionary treatment for patients suffering from
haematological diseases. Nonetheless, there are considerable challenges in the
implementation of these therapies in this rapidly evolving eld. These challenges
include but are not limited to the complexity of the supply chains for these living
drugs and the management of side effects, requiring centre qualication as well as
additional and ongoing education of health care professionals; the long-term fol-
low- up of patients treated with therapies with curative intent; the myriad regulatory
requirements at the European Union and local level; and reimbursement of the treat-
ments by budget-constrained authorities.
The challenges in the eld of cellular therapies require cross-stakeholder col-
laboration, including patient representatives, health care professionals, pharmaceu-
tical companies, health authorities, health technology assessment (HTA) bodies and
reimbursement agencies, and medical organizations at both the European and
national levels. For these reasons, EBMT and EHA have launched the GoCART

222
Coalition, a multistakeholder initiative aiming to promote patient access to novel
cellular therapies manufactured from cells and tissues of haematopoietic origin and
to contribute to health and well-being through innovation via multistakeholder col-
laboration on clinical data, standards of care, education, and policy.
The aims of the GoCART coalition:
• Improve health outcomes for patients.
• Engage stakeholders and establish a sustainable European coalition in the eld
of cellular therapy.
• Collaborate and share data and knowledge to prevent duplication of effort and
maximize resources.
• Promote harmonization of data collection, education, standards of care, regula-
tory approval, and reimbursement processes in Europe.
• Set up a pre- and post-marketing registry that supports regulatory decision-
making and shared research purposes.
• Develop a cellular therapy education and information programme for patients
and health care professionals.
• Harmonize standards of care and centre qualication.
• Advance policies that further the shared mission and vision.
The coalition is open to all stakeholders relevant to achieving its mission. Both
institutional and individual members are invited to participate in work packages that
implement the Coalition’s mission, vision, and goals. Work package chairs are
accountable to an executive committee with a balanced representation of stakehold-
ers, and the executive committee functions as the primary decision-making body
and determines the overall strategy of the coalition.
The following content work packages have been created:
1. Data harmonization
a. Context: In Europe, clinical data from patients treated with gene and cellular
therapies are reported to many registries, each built for a limited purpose,
with different governance rules and specic software tools managing the
data. This results in siloed data, inefciencies, and duplication of efforts.
b. Overall aim: Create a central EU data registry for harmonized collection of
clinical data on patients treated with cellular therapies to support collabora-
tive studies and regulatory decision-making.
2. Standards of care
a. Context: Gene and cellular therapies are inherently complex products, and
treatment administration is restricted to qualied centres. With rapid develop-
ments and pending product approvals, there is a need to develop treatment
guidelines and harmonize centre qualication procedures across pharmaceu-
tical companies, accreditation bodies, and national requirements.
b. Overall aims: (1) To develop harmonized guidelines on patient and product
management for health care professionals; (2) to reduce inspection burden
and redundancies by developing and implementing consensus-driven
S. R. Terwel et al.

223
requirements and qualication standards for clinical teams delivering gene
and cellular therapies from cells and tissues of haematopoietic origin.
3. HTA process
a. Context: Health technology assessment bodies and reimbursement agencies
need to make decisions based on the best available estimates of the properties
and impact of new therapies. This is particularly challenging for gene and
cellular therapies, considering that authorizations may be based on small
patient groups, limited availability of (long-term) follow-up and comparator
data, the high costs of the products, and the increasing number of therapies on
the horizon. Although national procedures on health technology assessments
and reimbursement vary considerably, there is a common need for reliable
safety and effectiveness data.
b. Overall aim: Leveraging the central registry for gene and cellular therapy as
a suitable data source for health technology assessment.
4. Education
a. Context: Gene and cellular therapies are complex products that require com-
prehensive and ongoing training of health care professionals as well as
patients and caregivers. A plethora of training courses are already offered by
MAHs as well as health organizations, which can lead to considerable
overlap.
b. Overall aim: Develop harmonized educational programmes for different
groups of health care professionals and patients.
5. Policy and advocacy
a. Context: Gene and cellular therapies are subject to EU and national regula-
tions affecting their preparation, administration and patient access. These
new therapies challenge these regulations, which were designed for more tra-
ditional pharmaceutical products, and health authorities are assessing how
they will adapt.
b. Overall aim: Represent and promote the interests of the GoCART coalition
and its stakeholders in EU policy-making by engaging with EU institutions
and other relevant stakeholders.
6. Scientic excellence
a. Context: Scientic research on gene and cellular therapies has increased
substantially in recent years. With real-world data becoming increasingly
available, many scientic questions can be explored from different per-
spectives. Only by working together can we leverage enough data to con-
duct meaningful research. GoCART wants to maximize the use of data
collected in the central registry as well as data available to other stakehold-
ers and to facilitate further collaboration between stakeholders. While
strongly protecting condentiality, the guiding principle should be ‘collect
once, use often’ to advance our knowledge in the eld of gene and cellular
therapies, support better decision- making, and drive efciencies for all
stakeholders.
b. Overall aim: Stimulate scientic discussion across stakeholders, facilitate the
setup of joint research projects, and avoid duplication of scientic efforts.
43 GoCART

224
Take a look at our webpage for the most recent information: https://thegocartco-
alition.com
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
Key Points
• The GoCART cell coalition is a multistakeholder initiative in the eld of
cellular therapies for haematological disease.
• Stakeholders include patient representatives, health care professionals,
pharmaceutical companies, health authorities, health technology assess-
ment (HTA) bodies and reimbursement agencies, and medical organiza-
tions at both the European and national levels.
• The mission is to promote patient access to novel cellular therapies manu-
factured from cells and tissues of haematopoietic origin and to contribute
to health and well-being through innovation via multistakeholder collabo-
ration on clinical data, standards of care, education, and policy.
• The GoCART Coalition aims to achieve its mission through activities
organized in work packages on (1) data harmonization, (2) standards of
care, (3) HTA, (4) education, (5) policy and advocacy, and (6) scientic
excellence.
S. R. Terwel et al.

225
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_44
J. Snowden (*)
Department of Haematology, Shefeld Teaching Hospitals NHS Foundation Trust and
University of Shefeld, Shefeld, UK
e-mail: [email protected]
R. F. Duarte
Department of Hematology, Hospital Universitario Puerta de Hierro Majadahonda and
Universidad Autónoma de Madrid, Madrid, Spain
e-mail: [email protected]
44
Patient Referral
JohnSnowden andRafaelF.Duarte
Early and efcient patient referral is a critical step in the ability of potential candi-
dates to access CAR-T therapy. Despite improvements in centre qualication and
availability, regulatory and reimbursement frameworks, and addressing the educa-
tional needs of the various members of the health care team, referring haematolo-
gists and oncologists identify major barriers to prescribing CAR-T therapy,
including cumbersome logistics, high cost and toxicity, and clinical challenges,
such as deterioration of the patient prior to CAR-T administration and the need for
bridging chemotherapy while awaiting manufacturing (Chavarría 2021).
Pathways for referral vary between countries and regions, but generally, patients
are referred to the regional CAR-T specialist multidisciplinary team (MDT) accord-
ing to agreed pathways, which in turn, may be linked with national committees
often necessary for additional clinical support and/or to conrm funding. These
specialist MDTs conrm patient eligibility in line with the manufacturer’s licence
and based on diagnosis, age, tness, disease, and treatment stage. Thereafter, the
CAR-T centres will arrange to assess the patient directly (with their carers) and
provide detailed information enabling the patient to understand the potential bene-
ts, risks, and complications of treatment and to provide informed consent.
Irrespective of the treatment site, clinicians must consider the eligibility of
potential patients for CAR-T cells at an early stage so that strategic decisions can be
made regarding the best therapeutic pathway. Eligibility should be directly con-
rmed with regard to age, tness, disease, and treatment stage. In addition, referring

226
clinicians should inform their patients of the potential of using CAR-T cells in their
treatment early in the pathway, especially if treatment may take place in another
centre some distance from their home or base centre (Gajra et al. 2020). In addition
to conrmation of eligibility and logistical arrangements with the treatment site,
prompt referral and good communication are also desirable to plan the salvage pro-
tocol for bridging CAR-T therapy, particularly because dened recovery periods
may be required before leukapheresis and the quality of circulating T-cells may
decrease with increasing chemotherapy exposure. Sometimes patients without a
high peripheral disease burden and sufcient circulating T-cells (e.g., total lympho-
cyte count of >0.5 × 10
9
/L or a peripheral blood CD3 count of >150 per μl) may be
able to undergo leukapheresis for CAR-T cells before starting salvage therapy for
relapse. For other patients, planning bridging therapy with the CAR-T therapy cen-
tre will be necessary. Therapies likely to signicantly impair lymphocyte number
and/or function should be avoided to allow successful leukapheresis for CAR-T cell
therapy. Therefore, careful scheduling and prioritization of patients is required,
including planning for leukapheresis, particularly given that CAR-T manufacture
can take over one month. Finally, capacity planning is required for subsequent
stages of care in the CAR-T centre, and later, shared care arrangements will enable
continuity of care after a patient returns home (Maus and Levine 1996).
References
Chavarría T. Real-world regulatory issues in the implementation of advanced therapies.
Multistakeholder forum at 47th EBMT annual meeting. 2021. Available at www.ebmt.org
Gajra A, Jeune-Smith Y, Kish J, Yeh T-C, Hime S, Feinberg B.Perceptions of community hema-
tologists/oncologists on barriers to chimeric antigen receptor T-cell therapy for the treatment of
diffuse large B-cell lymphoma. Immunotherapy. 2020;12(10):725–32. https://doi.org/10.2217/
imt- 2020- 0118.
Maus MV, Levine BL.Chimeric antigen receptor T-cell therapy for the community oncologist.
Oncologist. 1996;21:608–17. https://doi.org/10.1634/theoncologist.2015- 0421.
Key Points
• Prompt early patient referral from the base hospital to the treatment centre
facilitates various aspects of the planning for CAR-T therapy.
• The learning curve in the CAR-T therapy framework will also inform and
facilitate the management and referral of patients for other advanced ther-
apy medicinal products.
J. Snowden and R. F. Duarte

227
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
44 Patient Referral

229
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_45
C. Haag (*)
University Hospital Carl Gustav Carus Dresden, Dresden, Germany
e-mail: [email protected]
45
Treatment Coverage
andReimbursement
CornelieHaag
The conditions for reimbursement for CAR-T cell therapy are not uniform in
Europe. Most European countries use a DRG system for billing hospital services,
but the details vary. Nonetheless, the similarity is that expensive therapies, such as
CAR-T cell therapy, are initially not included in the DRG system. Most countries
possess instruments to ensure the nancing of such expensive therapies outside the
DRG system as separate payments. These reimbursement instruments of DRG sys-
tems are used in most countries both for short-term nancing for innovative and new
therapies and as long-term additional fees within the respective DRG system.
Individual countries maintain different regulations, and therefore, hospitals have the
responsibility to determine the specic requirements of their country before estab-
lishing CAR-T cell therapy.
One should consider that other signicant costs exist in addition to the price of
the actual CAR-T cell product, which has been agreed upon with the pharmaceuti-
cal industry. In addition to the usual hospitalization costs, the price of the inpatient
stay for the administration of CAR-T cells can include the costs for intensive care
and expensive medication, such as tocilizumab. These additional costs are generally
reimbursed through the established system in each country. However, at least 2
years are required to integrate the costs of a new therapy or method into the
existing DRG.
The special feature of CAR-T cell therapy is that the hospital needs to collect
lymphocytes from the patient in advance through apheresis. This initial product for
the production of CAR-T cells induces further costs that are usually not reimbursed.
The implementation of this new therapy in a hospital should not be underesti-
mated. In addition to the training of staff for this new type of therapy, high demands
are placed on quality management by both the pharmaceutical industry and the
government. These structural costs (mostly personnel costs) for the hospital must be

230
agreed upon separately with health insurance companies or the government, depend-
ing on the state-dependent reimbursement system.
A single hospital has a minor impact on the pricing of a CAR-T cell product; this
is usually done by negotiation between pharmaceutical companies and government
agencies.
In addition to the reimbursement of the CAR-T cell product at the price set by
these negotiations, the additional costs of this therapy are reimbursed differently,
particularly within Germany. Efforts are being made to centralize these negotia-
tions, but the success of such a centralized negotiation depends on the structures and
organization of the numerous health insurance companies in Germany.
In Germany, the individual hospital then becomes responsible for the specic
reimbursement of costs for each individual patient. In the case of extremely high
costs, advanced agreements are usually made between the health insurance and the
hospital.
Before initiating CAR-T cell therapy, every doctor or hospital should be aware of
the different regulations in each country to avoid not receiving reimbursement for
this expensive therapy.
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
Key Points
• Different rules in different countries.
• Additional costs aside from the cost of the CAR-T cell product.
C. Haag

231
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_46
M. Abou-el-Enein (*)
Division of Medical Oncology, Department of Medicine, and Department of Stem Cell
Biology and Regenerative Medicine, Keck School of Medicine, University of Southern
California, Los Angeles, CA, USA
Joint USC/CHLA Cell Therapy Program, University of Southern California, and Children
Hospital, Los Angeles, CA, USA
e-mail: [email protected]
J. Gauthier
Clinical Research Division, Fred Hutchinson Cancer Research Center, Seattle, WA, USA
Division of Medical Oncology, University of Washington, Seattle, WA, USA
e-mail: [email protected]
46
The Value ofCAR-T-cell Immunotherapy
in Cancer
MohamedAbou-el-Enein andJordanGauthier
The development of genetically modied chimeric antigen receptor (CAR) T-cells
to target cancer by conferring tumour antigen recognition has tremendously
improved the ght against the disease and broadened treatment options for haema-
tological malignancies (Elsallab etal. 2020b). However, in contrast to conventional
drugs that patients can easily access, the implementation of CAR-T-cell therapy in
routine clinical practice poses signicant challenges. Access to CAR-T-cell prod-
ucts is currently limited to specic certied centres meeting the requirements set up
by manufacturers and regulatory agencies. There are also issues regarding insurance
coverage, reimbursement, affordability, and pricing, which have critical impacts on
broadening patient access to these novel therapies (Abou-El-Enein etal. 2016a, b).
Current list pricing ranges between $373,000 and $475,000 per one-time infusion
for the ve CAR-T-cell therapies currently approved by the FDA (tisagenlecleucel,
Kymriah
®
; axicabtagene ciloleucel, Yescarta
®
; brexucabtagene autoleucel,
Tecartus
®
; lisocabtagene maraleucel, Breyanzi
®
; idecabtagene vicleucel, Abecma
®
).
In addition to the cost of the CAR- T- cell product, patient preparation (leukapheresis
and/or lymphodepletion), product infusion, pre- and post-infusion patient manage-
ment, and monitoring for side effects (Wagner etal. 2021) signicantly add to the
nal price tag. There are calls for restructuring the current payment and

232
reimbursement models to allow better access to CAR-T-cell therapies (Abou-El-
Enein et al. 2014). However, this would only be possible after examining the
strength of clinical evidence generated during product development (Abou-El-
Enein and Hey 2019; Elsallab etal. 2020a) and, most importantly, by determining
the value of CAR-T-cell therapy.
Efcacy does not automatically entail value. Quality-adjusted life years (QALYs)
per dollar spent reect a well-accepted measure of cost-effectiveness to assess
value. QALYs enable evaluation of the impact of a certain therapy on the entire
lifespan of a patient (quantity of life) and on health-related quality of life (HRQoL),
reecting a main parameter of treatment outcomes (Whitehead and Ali 2010). As
composite estimates of mortality and morbidity, QALYs are conventionally calcu-
lated by accumulating life years attained from a utility value specic to certain
health states. Preference elicitation studies in patients with a certain medical condi-
tion, such as in clinical trial scenarios, or in the general population serve as the basis
to derive this utility value (Prieto and Sacristán 2003; Whitehead and Ali 2010;
Sanders etal. 2016; Fiorenza etal. 2020).
Various models have been utilized to assess the cost-effectiveness of CAR-T-cell
therapy. With respect to Kymriah
®
and Yescarta
®
, Lin etal. used a decision analytic
Markov model and data from multicentre single-arm trials from a US health payer
perspective for patients with relapsed or refractory (r/r) adult large B-cell lym-
phoma. CAR-T-cell therapies were compared to salvage chemotherapy and stem
cell transplantation by incorporating certain assumptions regarding long-term effec-
tiveness in the model. Yescarta
®
was shown to prolong life expectancy by 8.2 years
at $129,000/QALY gained (95% uncertainty interval, $90,000 to $219,000) when
assuming a 40% 5-year progression-free survival (PFS). Kymriah
®
led to an increase
of 4.6 years at $168,000/QALY gained (95% uncertainty interval, $105,000 to
$414,000/QALY) when assuming a 35% 5-year PFS (Lin etal. 2019). The study
indicated that lowering the list price of Yescarta
®
and Kymriah
®
to $250,000 and
$200,000in the US, respectively, or implementing payment only for an initial com-
plete response (at current prices) would enable both CAR-T-cell therapies to cost
less than $150,000/QALY even at the more conservative assumption of a 25%
5-year PFS (Lin etal. 2019). Using data of paediatric patients with r/r B-cell ALL,
Sarkar et al. built a microsimulation model to measure the incremental cost-
effectiveness ratio (ICER) (Sanders etal. 2016) comparing CAR-T-cell therapy to
standard of care, considering ICERs below a threshold of $100,000 per QALY as
cost-effective (Sarkar etal. 2019). Assuming a 76% 1-year survival, they demon-
strated an increase in overall cost by $528,200 with improved effectiveness by 8.18
QALYs, leading to an ICER of $64,600/QALY. However, if the assumption was
modied to 57.8% 1-year survival, CAR-T-cell therapy in paediatric B-ALL patients
was no longer cost-effective. While probabilistic sensitivity analysis showed CAR-
T- cell therapy to be cost-effective in approximately 95% of iterations at a level of
willingness to pay $100,000/QALY (Sarkar etal. 2019), assumptions made regard-
ing long-term outcomes in both models need to be conrmed by real-world data
with longer follow-up duration to enable robust validation of study outcomes.
M. Abou-el-Enein and J. Gauthier

233
When discussing value-based considerations, social value gained by CAR-T-
cell therapy in the long term should also be taken into account. Offering a cure
to paediatric cancer patients would enable them to lead a more productive life
(Fiorenza etal. 2020). Moreover, successful milestones reached with respect to
patenting (Jürgens and Clarke 2019) and regulatory and clinical success
(Elsallab etal. 2020b) will increase public recognition, nancial support, and
advancements in the entire cellular therapy eld. A recent study applied an eco-
nomic framework to measure the social value of CAR-T-cell therapy as a sum of
consumer surplus and prot for the manufacturing company (Thornton Snider
etal. 2019). Consumer surplus reected the difference between the added value
of health gains achieved by the therapy and its incremental cost, accounting also
for indirect costs and patient benets. The gained social value was determined
to be as much as $6.5 billion and $34.8 billion for paediatric ALL and DLBCL,
respectively, with a net social value gain of $952,991 per child treated for
B-ALL, even after including costs for production and treatment. However, they
also showed a critical effect of treatment delays that negatively affect the social
value generated by CAR-T-cell therapy, with a 1, 2, or 6 month treatment delay
leading to a 9.8%, 36.2%, and 67.3% loss of social value, respectively, for pae-
diatric ALL patients and a 4.2%, 11.5%, and 46.0% loss of social value, respec-
tively, for patients with DLBCL (Thornton Snider et al. 2019). Thus, timely
patient access is a key factor in the level of value achieved. Other key parame-
ters to optimize the value of CAR-T-cell therapies rely on improving response
rates, minimizing the risk of relapse and lowering the costs of toxicity manage-
ment (Fiorenza etal. 2020).
Although CAR-T-cell therapy is undoubtedly transforming the therapeutic land-
scape for cancer patients, signicant economic challenges ought to be addressed to
allow broader and fairer access to these new therapies. Since most cost- effectiveness
models are highly assumption-sensitive, a longer follow-up duration is warranted to
better assess the value of CAR-T-cell therapies compared to alternative approaches.
Key Points
• CAR-T-cells have emerged as an important therapeutic approach for many
cancer patients; however, issues regarding insurance coverage, reimburse-
ment, affordability and pricing impact access to these novel therapies.
• Short-term clinical data have demonstrated the potential of CAR-T-cells to
become a cost-effective approach for cancer patients, but availability of
long- term clinical outcomes will be required to achieve this goal.
• The value of such a novel therapeutic modality should also be evaluated
within the social gains of cancer patients resuming normal and productive
lifestyles. However, these gains are dramatically inuenced by delays in
receiving the treatments.
The Value ofCAR-T-cell Immunotherapy in Cancer

234
References
Abou-El-Enein M, Hey SP. Cell and gene therapy trials: are we facing an “evidence crisis”?
EClinicalMedicine. 2019;7:13–4. https://doi.org/10.1016/j.eclinm.2019.01.015.
Abou-El-Enein M, Bauer G, Reinke P. The business case for cell and gene therapies. Nat
Biotechnol. 2014;32(12):1192–3. https://doi.org/10.1038/nbt.3084.
Abou-El-Enein M, Elsanhoury A, Reinke P.Overcoming challenges facing advanced therapies in
the EU market. Cell Stem Cell. 2016a;12:293–7. https://doi.org/10.1016/j.stem.2016.08.012.
Abou-El-Enein M, etal. Putting a price tag on novel autologous cellular therapies. Cytotherapy.
2016b;18(8):1056–61. https://doi.org/10.1016/j.jcyt.2016.05.005.
Elsallab M, Bravery CA, etal. Mitigating deciencies in evidence during regulatory assessments
of advanced therapies: a comparative study with other biologicals. Mol Ther. 2020a;18:269–79.
https://doi.org/10.1016/j.omtm.2020.05.035.
Elsallab M, Levine BL, etal. CAR-T-cell product performance in haematological malignancies
before and after marketing authorisation. In: The lancet oncology. Amsterdam: Elsevier;
2020b. p.104–16. https://doi.org/10.1016/S1470- 2045(19)30729- 6.
Fiorenza S, etal. Value and affordability of CAR-T-cell therapy in the United States. Bone Marrow
Transplant. 2020;55(9):1706–15. https://doi.org/10.1038/s41409- 020- 0956- 8.
Jürgens B, Clarke NS.Evolution of CAR-T -cell immunotherapy in terms of patenting activity. Nat
Biotechnol. 2019;37(4):370–5. https://doi.org/10.1038/s41587- 019- 0083- 5.
Lin JK, etal. Cost effectiveness of chimeric antigen receptor T-Cell therapy in multiply relapsed
or refractory adult large B-cell lymphoma. J Clin Oncol. 2019;37(24):2105–19. https://doi.
org/10.1200/JCO.18.02079.
Prieto L, Sacristán JA.Problems and solutions in calculating quality-adjusted life years (QALYs).
Health Qual Life Outcomes. 2003;1:80. https://doi.org/10.1186/1477- 7525- 1- 80.
Sanders GD, etal. Recommendations for conduct, methodological practices, and reporting of cost-
effectiveness analyses. JAMA. 2016;316(10):1093. https://doi.org/10.1001/jama.2016.12195.
Sarkar RR, etal. Cost-effectiveness of chimeric antigen receptor T-cell therapy in pediatric relapsed/
refractory B-cell acute lymphoblastic leukemia. J Natl Cancer Inst. 2019;111(7):719–26.
https://doi.org/10.1093/jnci/djy193.
Thornton Snider J, etal. The potential impact of CAR-T-cell treatment delays on society. Am J
Manag Care. 2019;25(8):379–86.
Wagner DL, etal. Immunogenicity of CAR-T cells in cancer therapy. Nat Rev Clin Oncol. 2021.
https://doi.org/10.1038/s41571- 021- 00476- 2.
Whitehead SJ, Ali S.Health outcomes in economic evaluation: the QALY and utilities. Br Med
Bull. 2010;96(1):5–21. https://doi.org/10.1093/bmb/ldq033.
Open Access
This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
M. Abou-el-Enein and J. Gauthier

235
© The Author(s) 2022
N. Kröger et al. (eds.), The EBMT/EHA CAR-T Cell Handbook,
https://doi.org/10.1007/978-3-030-94353-0_47
H. Schoemans (*)
Department of Hematology, University Hospitals Leuven, Leuven, Belgium
Department of Public Health and Primary Care, ACCENT VV, Katholieke Universiteit
Leuven- University of Leuven, Leuven, Belgium
e-mail: [email protected]
N. Bolaños
Lymphoma Coalition, Madrid, Spain
e-mail: [email protected]
L. Warwick
Lymphoma Coalition, Management, Mississauga, ON, Canada
e-mail: [email protected]
47
What do Patients Want? TheImportance
ofPatient-reported Outcomes
HélèneSchoemans, NatachaBolaños, andLornaWarwick
Understanding of what it means for patients to receive CAR-T therapy remains
insufcient due to the small number of studies with a quality of life (QOL) focus,
selection bias of respondents, high risk of attrition due to disease relapse, and lim-
ited length of follow-up. CAR-T therapy is often presented as a last option for
patients with advanced disease. The primary aim of the treatment is patient survival
and hopefully disease elimination. However, understanding other aspects of health,
such as functional status, cognitive function, psychosocial concerns, and other
health-related (QOL) issues, is key to appreciating the full impact of such therapies
at both the individual and societal levels.
Such information can only be accessed by asking patients and caregivers directly,
without going through the lter of a third party, using either patient-reported out-
come measures and/or qualitative methods, such as interviews or focus groups. This
approach is supported by the cell therapy community, but evidence remains limited
(Chakraborty etal. 2019; Shalabi etal. 2021).
Side effects, such as CRS, neurotoxicity, and B-cell aplasia, are well documented.
Importantly, many patients report other concerns that impact their well- being and
require appropriate support from their health care team (Bamigbola et al. 2021).

236
Hoogland and colleagues recently showed that over half of adult CAR-T recipients
complained of moderate to severe fatigue (84%), decreased appetite (73%), dry mouth
(61%), and insomnia (55%) in the rst 100 days following therapy, with a symptom
peak seen after approximately two weeks (Hoogland etal. 2021). Compared to base-
line, physical functioning signicantly improved, with decreased pain, fatigue, and
depression, but anxiety increased (Hoogland etal. 2021). In a follow-up study up to
1-year post-CAR-T cell infusion, approximately one-third of patients presented last-
ing moderate to severe fatigue and insomnia, and 20% had decreased memory com-
pared to baseline (Barata etal. 2021). In contrast, in children and adolescents (3–21
years) who had undergone CAR-T therapy for acute leukaemia, a steady signicant
improvement in QOL compared to baseline was seen from 3 months post-treatment in
all domains examined (Laetsch etal. 2019).
Mental health is a long-term issue, considering that up to 20% of 1 to 5-year
adult survivors reported clinically meaningful depression or anxiety and over one-
third experienced cognitive difculties (Ruark etal. 2020). Marziaz and colleagues
also showed meaningful improvement in QOL up to month 18 in all domains,
except for mental health (Maziarz etal. 2020). Of note, there have been no signi-
cant associations identied between the severity of CRS or ICANS and long-term
quality of life to date.
Little is known about patient priorities and needs after CAR-T therapy, but the cur-
rent literature underscores the importance of appropriate information. By interviewing
patients, Matthews etal. found that most felt unprepared for the emotional aspects of
CAR-T therapy nor were they prepared for the intensity of the toxicities (Matthews
et al. 2019). The importance of addressing issues, such as clear information on the
treatment trajectory (Bamigbola etal. 2021), nancial toxicity, and the importance of
family members and other caregivers, has also been described (Foster etal. 2020).
Future studies are needed to broaden the understanding of CAR-T cell therapy
survivorship to identify the themes most important to patients, potentially including
themes identied in other cell therapy recipients, such as impact on informal care-
givers, return to school/work, nancial issues, and access to care (Burns etal. 2018).
Outcome evaluation in large groups of patients with extended longitudinal follow-
up is particularly important to identify predictors of QOL, specically of mental
health and cognitive function, so patients undergoing CAR-T therapy can be better
informed and supported.
Key Points
• Symptom burden generally decreases over time, starting from 3 months
post CAR-T therapy.
• Mental health and cognitive function remain a concern in long-term
survivors.
• Currently, there is no indication of an association between CRS or ICANs
and long-term QOL.
• Patient priorities, expectations, and needs regarding CAR-T cell therapy
urgently need to be assessed.
H. Schoemans et al.

237
References
Bamigbola O, Dren N, Warwick L.Cross-sectional study of the side-effects prole and patient-
doctor communication about side effects in patients with lymphoma treated with CAR-T ther-
apy. Poster presented at: 3rd European CAR-T -cell Meeting 2021, Virtual, 2021.
Barata A, Hoogland AI, Hyland K, Kommalapati A, Irizarry-Arroyo N, Rodriguez Y, etal. Patient-
reported toxicities in axicabtagene ciloleucel recipients: 1-year follow-up. Transplant Cell
Ther. 2021;27(3):375.
Burns LJ, Abbetti B, Arnold SD, Bender J, Doughtie S, El-Jawahiri A, etal. Engaging patients in
setting a patient-centered outcomes research agenda in hematopoietic cell transplantation. Biol
Blood Marrow Transplant. 2018;24(6):1111–8.
Chakraborty R, Sidana S, Shah GL, Scordo M, Hamilton BK, Majhail NS.Patient-reported out-
comes with chimeric antigen receptor T cell therapy: challenges and opportunities. Biol Blood
Marrow Transplant. 2019;25(5):155–62.
Foster M, Fergusson DA, Hawrysh T, Presseau J, Kekre N, Schwartz S, etal. Partnering with
patients to get better outcomes with chimeric antigen receptor T-cell therapy: towards engage-
ment of patients in early phase trials. Res Involve Engage. 2020;6:61.
Hoogland AI, Jayani RV, Collier A, Irizarry-Arroyo N, Rodriguez Y, Jain MD, etal. Acute patient-
reported outcomes in B-cell malignancies treated with axicabtagene ciloleucel. Cancer Med.
2021;10(6):1936–43.
Laetsch TW, Myers GD, Baruchel A, Dietz AC, Pulsipher MA, Bittencourt H, etal. Patient-reported
quality of life after tisagenlecleucel infusion in children and young adults with relapsed or
refractory B-cell acute lymphoblastic leukaemia: a global, single-arm, phase 2 trial. Lancet.
2019;20(12):1710–8.
Matthews A, Sidana S, Seymour L, Pick N, Pringnitz J, Argue D, etal. QIM19-136: developing an
Ideal CAR-T cell therapy patient experience through human-centered design and innovation. J
Natl Compr Cancer Netw. 2019;17:3–5.
Maziarz RT, Waller EK, Jaeger U, Fleury I, McGuirk J, Holte H, et al. Patient-reported long-
term quality of life after tisagenlecleucel in relapsed/refractory diffuse large B-cell lymphoma.
Blood Adv. 2020;4(4):629–37.
Ruark J, Mullane E, Cleary N, Cordeiro A, Bezerra ED, Wu V, etal. Patient-reported neuropsychi-
atric outcomes of long-term survivors after chimeric antigen receptor T cell therapy. Biol Blood
Marrow Transplant. 2020;26(1):34–43.
Shalabi H, Gust J, Taraseviciute A, Wolters PL, Leahy AB, Sandi C, etal. Beyond the storm- subacute
toxicities and late effects in children receiving CAR-T cells. Clin Oncol. 2021;18(6):363–78.
Open Access
This chapter is licensed under the terms of the Creative Commons Attribution 4.0
International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you give appropriate
credit to the original author(s) and the source, provide a link to the Creative Commons license and
indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative
Commons license, unless indicated otherwise in a credit line to the material. If material is not
included in the chapter's Creative Commons license and your intended use is not permitted by
statutory regulation or exceeds the permitted use, you will need to obtain permission directly from
the copyright holder.
47 What do Patients Want? TheImportance ofPatient-reported Outcomes
